AWS Quantum Technologies Blog
Building advanced quantum chemistry calculations with Kvantify Qrunch on Amazon Braket
Quantum computing promises breakthroughs in chemistry simulations. However, building scalable calculations on today’s noisy quantum hardware remains challenging. This blog introduces Kvantify Qrunch software on Amazon Braket. It enables advanced chemistry calculations using efficient hybrid quantum algorithms on current Noisy Intermediate-Scale Quantum (NISQ) devices.
Chemistry is at the heart of several industries with profound economic and societal impact, such as pharmaceuticals and biotech. The quantum nature of molecular interactions presents a fascinating computational opportunity—one that positions this field as an ideal application area for quantum computing to create substantial value. However, the lack of efficient, chemistry-specific software that is relevant for noisy, large-scale devices impedes broad exploration, adoption, and deployment of quantum computing technology. Researchers and practitioners need software that abstracts away the complexity of designing quantum circuits and routing them to quantum computing hardware. Kvantify Qrunch addresses this need in the quantum computing software space, as illustrated in Figure 1.
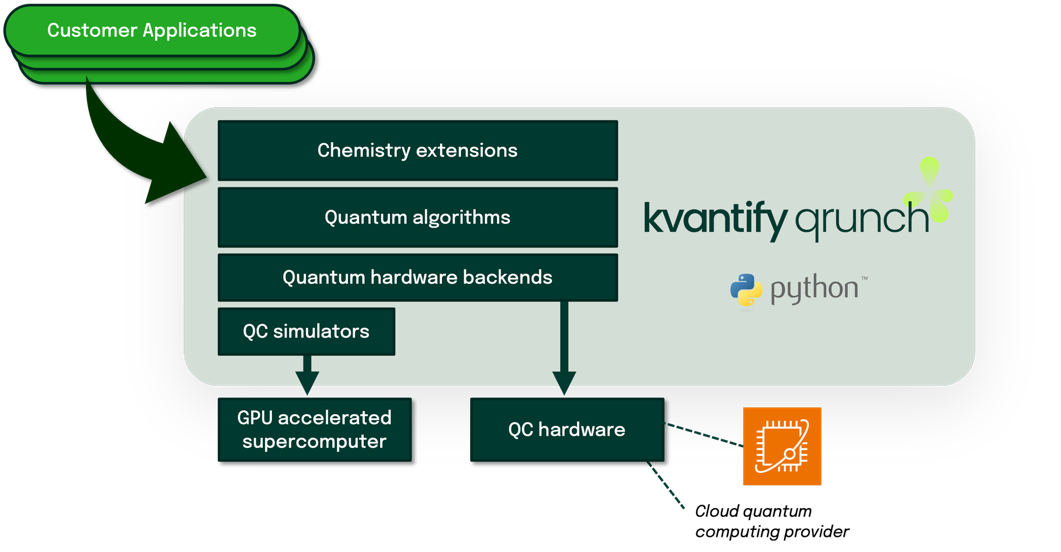
Figure 1 – Kvantify Qrunch integrates all the computational chemistry functionalities and manages the technical complexity of quantum computing, providing the missing software link to bridge the gap between advanced customer applications and execution on quantum computing hardware.
Getting started with Qrunch is simple. It is available here and a detailed technical documentation guides you in setting up and activating the license. From there, you can take advantage of several showcase applications and elaborate tutorials. Going through the tutorials you will learn how to set up advanced chemistry computations relevant to drug discovery, enzymatic reactions, and carbon capture, and run them on remote quantum computing hardware of your choice.
How Kvantify Qrunch makes it easy for chemists to adopt quantum computing
Kvantify Qrunch is tailored for professional computational chemists. Chemistry experts often lack quantum computing expertise or workflow experience. Qrunch removes these barriers. You can now build quantum applications without managing remote hardware backends. All that quantum computing know-how is built into Kvantify Qrunch, neatly stowed away under the hood, and made available to you in familiar Python form.
The following code snippets show you what working with Kvantify Qrunch looks like.
Flexible builder for chemistry problems
Advanced computational chemistry requires more than just a connection to a quantum computer. Qrunch integrates conventional chemistry methods as components that you can flexibly combine to build industry-relevant molecular electronic structure problems. The software guides you through defining their problem in a step-by-step fashion and ensuring that dependencies are matched to form valid configurations.
First, you specify the atomic configuration of the system, either by loading a .xyz file, as in the following example, or by directly specifying the individual atomic nuclei and their locations. Common file formats such as PDB can also be accommodated. If a reaction path problem is considered, a sequence of configurations is simply specified.
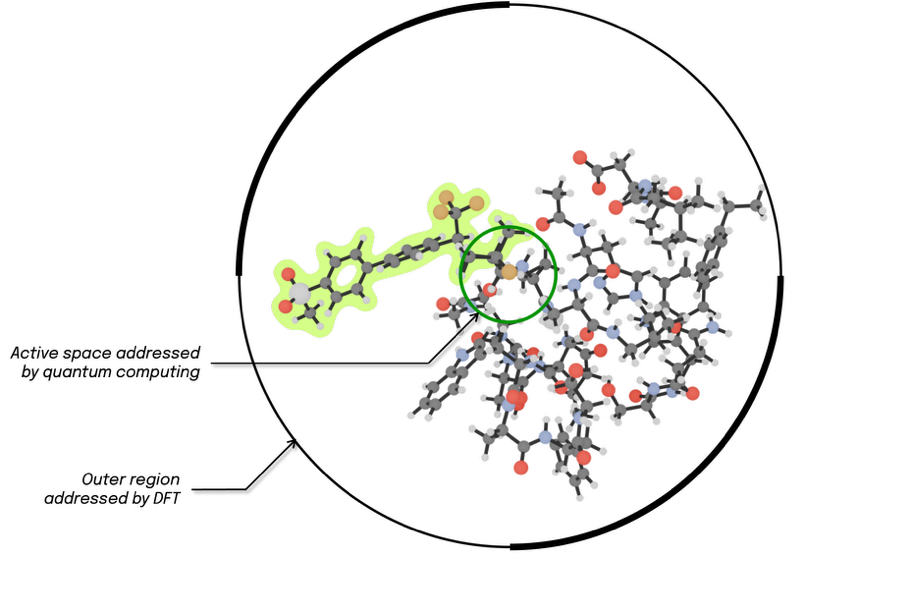
Figure 2 – Projective embedding allows a central region (encircled in green) of the molecular system to be addressed by quantum computing while lower-level methods are applied to surrounding layers. The illustration corresponds to the following drug discovery case presented with the structure highlighted in green being the odanacatib ligand binding covalently to the larger Cathepsin-K enzyme.
You can embed your system into an environment using the projective embedding technique. This lets you apply different methods to different regions. During configuration, specify which atoms belong to the quantum calculation. Think of this as an onion-like structure. Apply quantum computing to inner regions where, for instance, static electronic correlations matter. Use Density Functional Theory (DFT) for outer regions. Optionally, add molecular mechanics for the outermost layer. This selective approach optimizes spending of resources, exploiting quantum computing in demanding regions where classical methods struggle, while enabling large systems to be handled.
import qrunch as qc
file_path = “path_to_your_molecular_structure.xyz”
config = qc.build_molecular_configuration(
file_path,
basis_set="sto3g",
embedded_atoms=[0,1,2],
charge=0,
spin_difference=0
)
The system is by default configured as a Full Configuration Interaction (Full-CI) problem, which in most cases is intractable. It is therefore necessary to modify it by introducing an active space for the region treated by quantum computing.
problem_builder = (qc.problem_builder_creator()
.ground_state()
.projective_embedding()
.add_problem_modifier()
.active_space(
number_of_active_spatial_orbitals=26,
number_of_active_alpha_electrons=13,
)
.create()
)
problem = problem_builder.build_restricted(config)
As the preceding examples show, molecular problems can be constructed with just a few lines of code.
Setting up the quantum algorithms and executing the calculation
Kvantify Qrunch includes Kvantify’s proprietary algorithms, FAST-VQE, and BEAST-VQE. These are highly efficient operator selection strategies particularly well-suited for ground state energy calculations. Here, operators are adaptively added to a quantum circuit that iteratively approaches a representation of the distribution of Pauli-strings corresponding to the target ground state. At each iteration, the current quantum circuit is executed and the output bitstring resulting from the qubit measurements correspond to a sample drawn from the intermediate distribution of Pauli strings encoded by the circuit. Quantum computing is thus used to extract information about this highly complex distribution, feeding into a metric that guides the selection of the next operator to be added to the circuit. The metric and subsequent energy evaluation is carried out on a classical computer. While this is specifically a limitation of hybrid algorithms, we believe that there will always be a classical compute workload associated with quantum computing. In developing Qrunch, we have optimized the classical computations, associated with FAST-VQE and BEAST-VQE, by orders of magnitude, essential for scaling up the quantum computing part of the workflow. The FAST-VQE algorithm has been presented in more detail in a previous blog post.
While FAST-VQE considers both single and double electronic excitations, BEAST-VQE is an approximate approach that considers only paired electron excitations and treats them as bosonic particles. This approximation delivers three key benefits. First, it halves the number of required qubits. Second, it reduces quantum circuit complexity. Third, it re-groups the Hamiltonian to reduce measurements to a constant. In comparison, the number of measurements required for vanilla VQE scales as N4, where N describes the size of the system. This makes BEAST-VQE highly scalable and cost-efficient – advantages that are crucial for solving industry-relevant problems on current noisy quantum hardware. In addition, both FAST-VQE and BEAST-VQE exhibit noise-resilience due to the common sampling strategy.
With Kvantify Qrunch, setting up the quantum computing part is as easy and flexible as configuring the chemistry of the problem. The software supports IDE autocompletion and the fluent builder pattern ensures that decisions are made in a consistent and valid manner.
The following code example sets up a quantum computation for gate selection based on BEAST-VQE using a sampler with 1000 circuit shots as input.
shots = 1000
sampler = qc.sampler_creator().excitation_gate().create()
gate_selector = (
qc.gate_selector_creator()
.fast()
.with_shots(shots)
.with_sampler(sampler)
.with_heuristic_gradient()
.create()
)
calculator = (
qc.calculator_creator()
.vqe()
.iterative()
.beast()
.with_gate_selector(gate_selector)
.create()
)
result = calculator.calculate(problem)
Various choices of backends
Kvantify Qrunch seamlessly enables you to execute the configured problem on a selection of different backends. Facilitating efficient development of industry-relevant quantum computing applications, Qrunch uses a built-in high-performance simulator capable of simulating problems up to 30 qubits on a standard laptop in roughly 1 second. This is possible through a combination of exploiting knowledge about the structure of chemistry problems and efficient memory usage when representing quantum states.
An equally important facilitator of quantum computing uptake is the simplicity with which you can delegate execution to quantum computing hardware in the cloud. Kvantify Qrunch comes with pre-established interfaces to Amazon Braket’s diverse gate-based quantum hardware, including superconducting and trapped-ion quantum computers from providers like Rigetti, IQM, and IonQ. As an example, running a computation on the Rigetti Ankaa-3 hardware via Amazon Braket can be achieved by the few lines of code:
from braket.devices.devices import Devices
sampler = (
qc.sampler_creator()
.backend()
.choose_backend()
.amazon_braket(device=Devices.Rigetti.Ankaa3)
.create()
As the quick walk-through illustrates, this can help incorporate a quantum workflow into a larger classical computation. In short, Kvantify Qrunch allows chemists to focus on their domain expertise while seamlessly making quantum computing available as a new, valuable resource, and enabler for innovation.
Drug discovery showcase demonstrates the scalability
Now we walk through a concrete example using Qrunch. The example demonstrates three capabilities: problem scaling, quantum hardware utilization, and integration into industry-relevant chemistry workflows.
Covalent ligands are a fascinating and powerful class of drugs that bind to their biological targets by forming a covalent bond, well-known examples being aspirin, and penicillin. This type of binding offers several compelling advantages: enhanced selectivity, prolonged duration of action, and improved potency compared to non-covalent drugs. However, this power comes with a challenge: such strong interactions also run the risk of off-target effects and unintended chemical reactions, which can lead to adverse outcomes.
From a computational perspective, modeling covalent binding is a difficult task. Most conventional tools, such as force fields, often fall short when it comes to simulating bond formation or breaking. This limitation stems from how such empirical approaches work. They rely on pre-fitted parameters derived from existing molecular configurations. These parameters cannot capture complex quantum mechanical changes during chemical transformations. Electron distributions shift and new bonding patterns emerge in ways the parameters cannot predict. Quantum chemistry methods can overcome these limitations and provide crucial insight into drug-binding mechanisms by directly modeling the quantum mechanical foundations of molecular interactions. Quantum computing, in principle, may unlock the computational power needed to scale quantum chemistry simulations, allowing researchers to model intricate binding mechanisms and molecular transformations with enhanced accuracy and detail that drug discovery demands.
A case relevant to osteoporosis treatment
The Cathepsin-K enzyme is a protease involved in breaking down bone and cartilage and is actively studied as a therapeutic target for postmenopausal osteoporosis. The odanacatib ligand is a potent and highly selective covalent inhibitor of Cathepsin-K, and the resulting complex is the target case. The quantum computing region is defined by two atoms. The first is the electrophilic carbon from the ligand’s nitrile functional group. The second is the nucleophilic sulfur atom in Cathepsin-K’s Cys-25 Cysteine residue. They localize the active space to which orbitals are assigned. The surrounding environment of these active orbitals is modelled with projective embedding, where Kohn-Sham DFT provides the methodology to incorporate environmental effects into the core active region. The pool of all pair electron excitations will then be generated from this active space and will be added via the gate selection procedure of the BEAST-VQE algorithm on a quantum device or, alternatively, on a simulator.
Charting the path to 100 qubits
We executed the problem on different backends, going from state vector simulations to real quantum hardware. Beyond 30 qubits resolution of the problem the computation time of noisy simulations become unfeasible. To scale further, it is necessary to execute computations on real quantum hardware. For this task, we used the Rigetti Ankaa-3 device, available via Amazon Braket. As previously explained, VQE algorithms involve classical computations for certain tasks, which were executed with our exact chemistry-optimized simulator. As we scaled the problem to larger qubit numbers, this task was found to be a bottleneck – the classical computational workload increases dramatically as the size of the state vector grows. To address this challenge and enable full exploitation of Ankaa-3, Kvantify’s memory-restricted simulator was deployed. It retains a user-specified number of state vector components, limiting memory usage, and enabling simulation of systems of up to 100s of qubits. Although this introduces an approximation, the impact on the final energy accuracy is minimal when a reasonable state vector pruning is chosen.
This enabled us to fully leverage the Ankaa-3 hardware to perform a calculation on 80 qubits, specifically within an active space AS(80, 24) with 80 orbitals and 24 electrons. The corresponding convergence plot for a single-point calculation of the reaction is shown in Figure 3, together with two benchmark calculations: a state vector simulation (SV) and a random sampling simulation. These two extremes represent, respectively, the cases of noiseless hardware and completely noise-dominated hardware performance. The very encouraging observation is that the calculation performed on Ankaa-3 closely follows the state vector simulation, demonstrating the algorithm’s scalability and noise resilience.
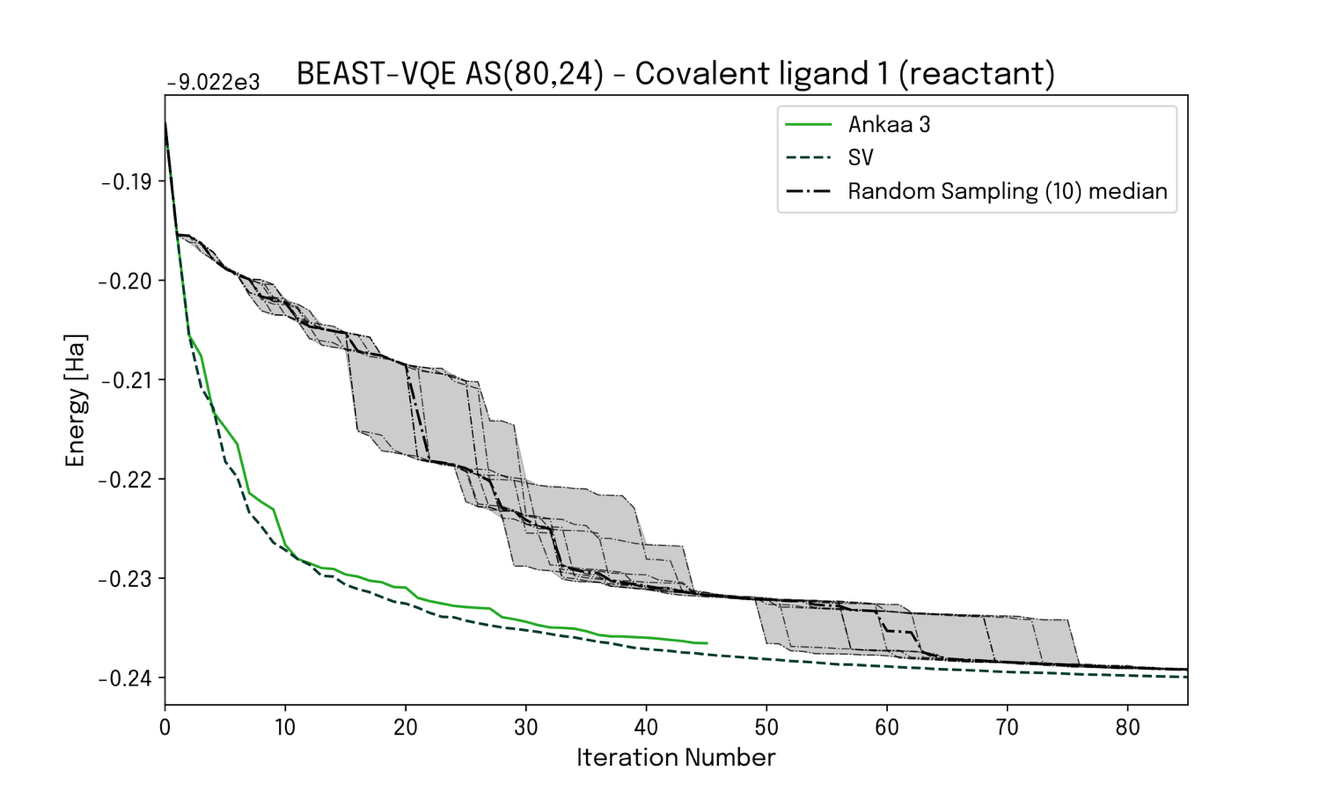
Figure 3 – Convergence plot for a single-point computation on 80 qubits executed on actual hardware (green), compared to benchmark computations corresponding to simulated noiseless (dashed) and completely noisy (dot-dashed; shaded region) hardware performance. The tendency of the hardware result towards the noiseless case demonstrates the ability of Kvantify Qrunch to fully exploit and extract close to optimal performance from current large-scale hardware.
This result is promising on two fronts. First, it shows that current noisy hardware can deliver high-quality samples of complex, large-scale distribution patterns. Second, it demonstrates the capabilities of Qrunch for real-world chemistry problems. Qrunch establishes complex chemistry workflows and delegates crucial aspects to quantum hardware. It uses scalable, noise-resilient algorithms to extract valuable results from hardware samples. Note, executing a computation on an active space of 80 orbitals occupied by 24 electrons is not feasible with conventional CI methods running on classical computers and would have to be targeted with approximate approaches.
Conclusion
With Kvantify Qrunch, NISQ hardware can become a utility for large-scale molecular simulations and development of advanced, scalable, and innovation-driving computational chemistry applications of quantum computers has moved a significant step closer. Now, professional chemists have a streamlined gateway to quantum computing, providing fully featured integration of computational chemistry methods, guided building of scalable hybrid quantum-classical problems, and a simple path to execution on cloud quantum computers as available through Amazon Braket. Mitigating the need for an expert background in quantum computing, Kvantify Qrunch enables everyone to exploit the computational capacity of current quantum computing hardware and expand the scale of chemistry simulations addressable by quantum computing. This encourages optimistic exploration of quantum computing and development of applications among chemistry professionals in industry and academia.
Reference
Kvantify Qrunch is available under three license options: Basic, Pro, and Enterprise versions. The Basic version is free to use and supports applications on up to 35 qubits on Kvantify partners’ hardware with access procured separately from the provider. The Pro version additionally supports an unlimited number of qubits and a wider range of quantum computing hardware and includes technical support. The Enterprise version includes all the capabilities of the Pro version and extended technical and scientific support.