亚马逊AWS官方博客
从代码到分子系列:一场由 AI 驱动的 EGFR 抑制剂发现之旅 — 深度融合 AWS Bedrock与 Claude Code/Claude Agent Skills,生命健康行业的科学活动探微
传统药物研发的”三座大山”
在深入介绍本文主题之前,让我们先直面药物研发领域的残酷现实:
- 时间成本:漫长的研发周期
-
- 从靶点发现到临床试验,一个新药的平均研发周期长达10-15年
- 早期药物发现阶段(靶点验证→先导化合物筛选→优化)就需要3-5年
- 化学家需要手动检索文献、下载数据、分析化合物性质、绘制结构图,每个步骤都是时间黑洞
- 经济成本:天文数字的投入
-
- 一个新药的平均研发成本高达26亿美元(数据来源:Tufts CSDD,2020)
- 其中约30%的费用花在早期药物发现阶段的”试错”上
- 大量资金浪费在低成功率的化合物筛选中(成功率不足5%)
- 技能壁垒:多学科交叉的复杂性
-
- 药物化学家需要掌握:化学信息学(RDKit)、分子对接(AutoDock)、数据分析(Python)、文献管理(EndNote)
- 不同工具之间数据格式不兼容,需要手动转换(如SDF→PDB→MOL2)
- 知识孤岛问题严重:化合物数据库(ChEMBL)、基因突变数据库(COSMIC)、文献数据库(PubMed)分散在不同平台,无法自动关联
从痛点到突破:Claude Agent Skills的解决方案
这正是本文的核心关注点:如何用一句提示词(prompt),让AI自动完成上述所有工作?
本文将详细记录如何驱动Claude Agent Skills完成一次完整的EGFR抑制剂药物发现流程——从数据库挖掘、结构-活性关系分析,到分子生成、虚拟筛选,再到文献综述和报告生成。传统流程需要反复切换工具、手动处理数据的”苦力活”,现在可以交给AI一键完成。
这不仅是一次技术实践,更是对AI如何革新科学研究范式的深刻思考。让我们开始这段旅程。
一、环境准备:核心工具介绍与安装
1.1 什么是Claude Code
Claude Code 是 Anthropic 推出的终端原生 AI 编程助手,基于 Claude 4 系列模型(如 Opus 4.5、Sonnet 4.5),专为代码全生命周期任务优化,可通过自然语言指令完成开发、调试、重构等工作,无需切换 IDE 或聊天窗口
Claude Code具有以下核心特点:
- 终端优先,无缝集成:直接运行在 Bash/Zsh 终端,深度适配 VS Code、JetBrains 等主流 IDE,遵循 Unix 哲学,支持管道、脚本化操作,可无缝嵌入 CI/CD 流程,减少环境切换成本。
- 超长上下文,全局理解:支持最高 200K tokens 上下文,能解析大型代码库的架构、依赖与逻辑,实现跨文件协同编辑,适配复杂项目开发。
- 全栈能力,自主执行:覆盖 40 + 编程语言,可根据需求生成代码、定位修复 Bug、重构优化、生成测试用例与 API 文档,还能执行 Git 操作、安全审查(如扫描 SQL 注入漏洞),自主完成开发任务。
- 安全可控,灵活扩展:修改文件需用户明确授权,适配团队编码规范;支持通过 SDK、GitHub Actions 扩展,可部署于 Amazon Bedrock 等平台,满足企业级安全与合规需求。
- 高效智能,性能领先:依托 Claude 4 系列模型的强推理能力,在 SWE-bench 测试准确率领先业界平均水平,能快速响应复杂开发需求,提升研发效率。
1.2 什么是Claude Agent Skills?
Claude Agent Skills是基于Anthropic公司的Claude AI模型,结合专业领域知识构建的技能系统。简单来说,它让AI不再是一个”万金油”的聊天机器人,而是能够调用专业工具、执行复杂任务的”科研助手”。
在药物发现场景中,claude-scientific-skills项目提供了140个即用型科学技能,涵盖多个生物医学领域:
- ChEMBL:访问全球最大的生物活性化合物数据库
- RDKit:进行化学信息学分析(分子描述符、SAR分析)
- Datamol:生成改进的分子类似物
- DiffDock:执行分子对接和虚拟筛选
- PubMed:检索生物医学文献
- COSMIC:查询癌症基因突变数据库
这些技能通过Claude Code的Plugin系统加载,Claude会自动识别任务需求并调用相应的工具。
1.3 如何使用Claude Code、Amazon Bedrock与claude-scientific-skills赋能药物研发全流程?
通过Claude Code +Amazon Bedrock 大模型服务+claude-scientific-skills的组合,可以利用专业领域知识、业界编码SOTA模型、长上下文、全栈代码能力,打通从生信分析、药物研发到临床与合规的药物研发全流程,尤其适配制药、生物技术公司的高复杂度、高合规性场景,例如:
- 生信分析自动化:支持单细胞测序(Scanpy/Scvi-tools)、基因组比对、GWAS、多组学整合,可一键生成预处理、差异分析、细胞注释、PPI / 细胞通讯分析代码,对接 GEO、ClinVar、COSMIC 等数据库,将数月流程压缩至分钟级。
- 药物研发全链路:从靶点发现、虚拟筛选(DiffDock、RDKit)、ADMET 预测(DeepChem)到分子优化,自动生成脚本并串联工具链;支持蛋白结构预测(AlphaFold)与设计,加速苗头化合物到候选分子迭代。
- HPC 与云原生适配:编写并行计算脚本、优化集群调度,适配大规模分子动力学、基因组组装等高性能任务,兼顾效率与成本控制。
- 临床与合规提效:自动生成 / 审查临床研究报告、试验方案、SOP,编码 GxP 合规的数据分析与可视化流程;对接电子数据系统,快速完成监管提交材料的数据清洗、统计与报告生成,如将临床报告周期从 12 周缩至 10 分钟。
- 科研与知识工程:批量检索 PubMed、OpenAlex 并生成综述、引用格式;对接内部知识库,实现试验结果、标准流程的秒级检索与问答,支撑研发决策。
1.4 环境准备
使用前需要安装配置Claude Code和Amazon Bedrock环境,并下载并加载claude-scientific-skills,步骤如下:
- 安装Claude Code
-
- 创建Bedrock API key
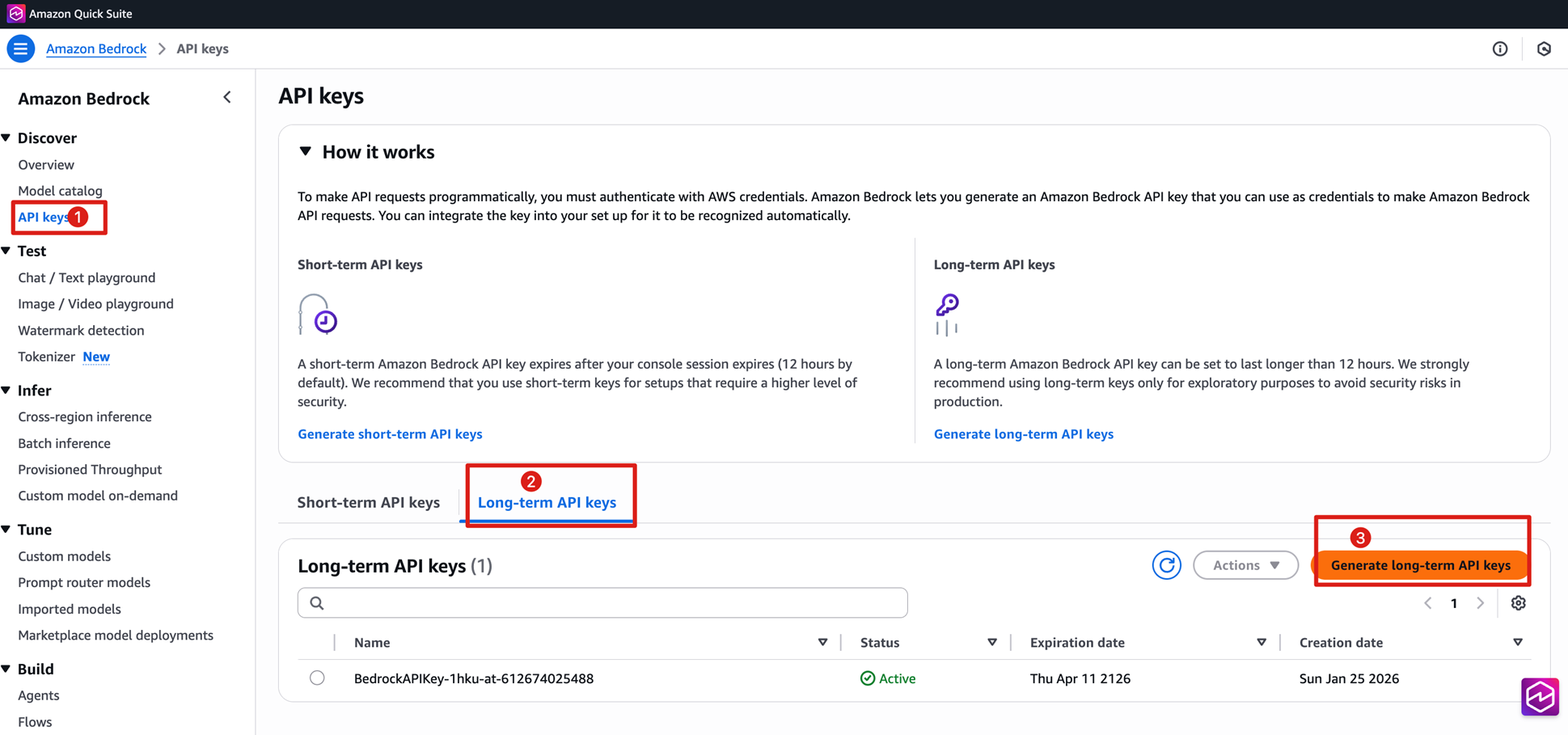 |
- 配置环境变量
- 打开claude code
- 在claude code中安装Claude Scientific Skills
注:本文实践使用的是Amazon Bedrock上的Claude Sonnet 4.5模型。
1.5 EGFR抑制剂药物发现工作流程演示介绍
启动工作流
只需在Claude Code对话框输入你的科研需求。本次工作流的提示词为:
"尽可能使用你能访问的所有技能。从ChEMBL查询EGFR抑制剂(IC50 < 50nM),用RDKit分析构效关系,用datamol生成改进的类似物,用DiffDock针对AlphaFold的EGFR结构进行虚拟筛选,在PubMed搜索耐药机制,检查COSMIC数据库中的突变,并创建可视化图表和综合报告。"
这段话包含了8个连续任务,传统方式需要人工切换多个软件工具,耗时数天。但Claude Agent Skills已经包括了常用的生物信息,计算化学,数据分析等技能,不需要额外安装,而且还能自动编排这些任务,在1小时内完成全部流程。
流程图示意如下:
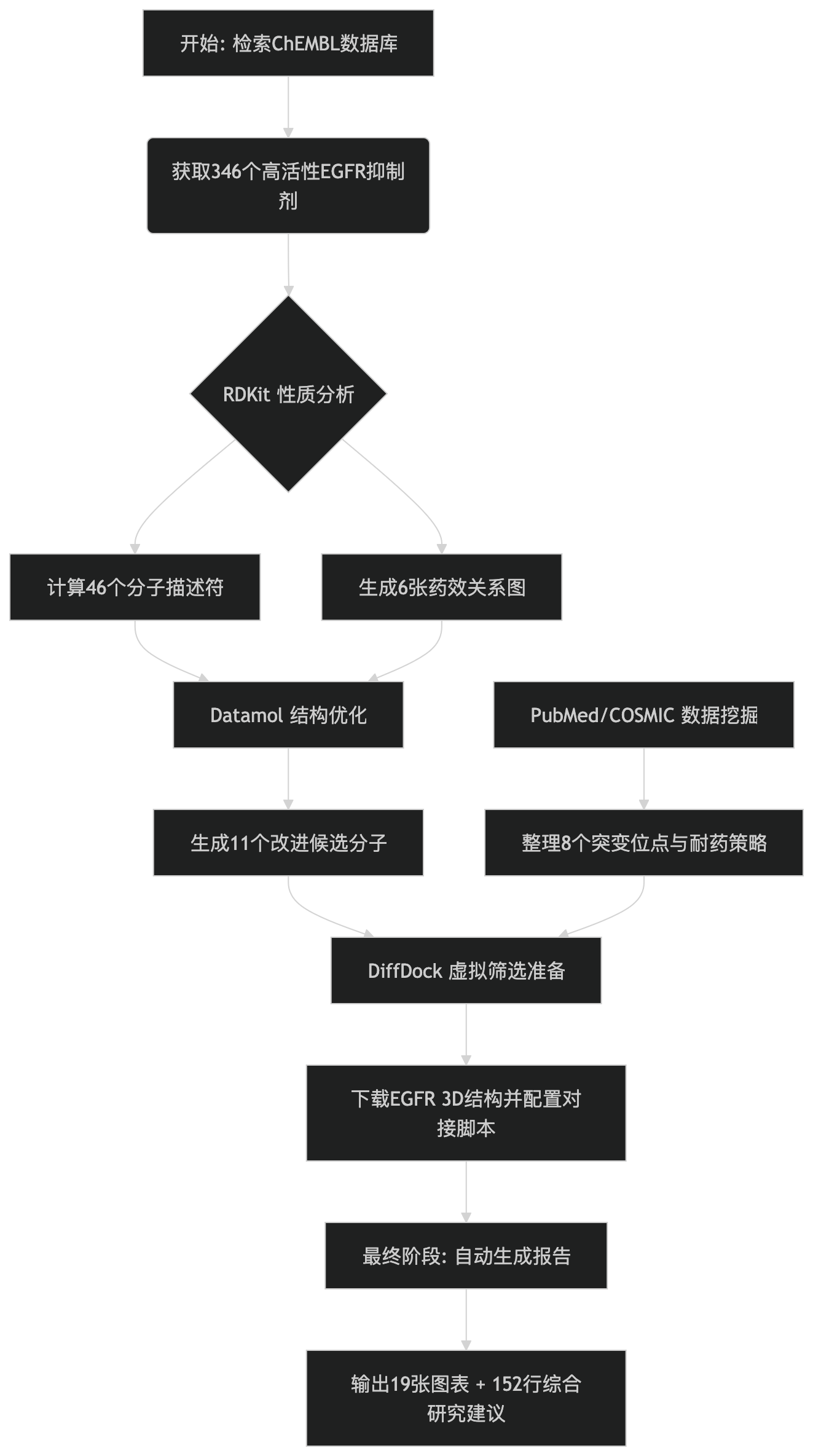 |
自动化执行与结果输出
当你按下回车键后,Claude开始自动执行这8个任务:
- 检索化合物数据库(ChEMBL)
从全球最大的生物活性化合物数据库检索出346个高活性EGFR抑制剂(IC50 < 50 nM)——找到精准的”分子钥匙”。
- 计算分子性质(RDKit)
计算分子量、脂溶性、极性等46个描述符,自动生成6张图表,揭示”分子结构”与”药效强度”的关系。
- 生成改进分子(Datamol)
基于构效关系分析,对高活性分子进行原子替换和结构优化,生成11个”变体”候选药物。
- 虚拟筛选准备(DiffDock)
下载EGFR蛋白3D结构,准备分子对接脚本和输入文件,配置好对接参数供后续GPU环境运行。
- 文献与突变数据挖掘(PubMed & COSMIC)
检索25篇耐药机制文献,整理8个关键突变位点(T790M、C797S等)的频率和治疗策略。
- 自动生成报告
输出12张分子结构图、6张性质分布图、1张散点图,以及152行综合报告(含研究背景、数据分析、设计建议)。
二、科研实践:从数据挖掘到分子设计
本章节围绕 EGFR 抑制剂药物设计,通过四步构建完整科研流程:先挖掘高活性化合物奠定基础,再经 SAR 分析揭示构效规律,接着生成优化候选分子,最后准备虚拟筛选验证。四步层层递进,从数据获取到规律解析、分子设计再到验证准备,既解决了药物研发数据分散、设计盲目等问题,又确保了候选药物的活性与可行性,展现了 AI 驱动药物发现的核心逻辑闭环。
2.1 数据挖掘:346个高活性EGFR抑制剂
这是药物发现的起点,需要从海量化合物中找到”有效的分子”。
这会缩短筛选时间,提高命中率,节省成本。
背景知识:表皮生长因子受体(EGFR)是非小细胞肺癌(NSCLC)中最重要的治疗靶点之一。EGFR突变存在于约15%的NSCLC患者中,尤其在亚洲人群中更为常见。针对EGFR的酪氨酸激酶抑制剂(TKIs)如厄洛替尼、吉非替尼已成为一线治疗药物。
1. Zhang YL, et al. Oncotarget. 2016;7(48):78985-78993.
2. Shi Y, et al. J Thorac Oncol. 2014;9(2):154-162.
3. Batra U, et al. BMJ Open Respir Res. 2023;10(1):e001492.
4. Rosell R, et al. Lancet Oncol. 2012;13(3):239-246.
5. Mok TS, et al. N Engl J Med. 2009;361(10):947-957.
Claude首先从ChEMBL数据库检索了IC50 < 50 nM的EGFR抑制剂,共计346个化合物。这些化合物的活性范围从0.006 nM到49 nM,涵盖了从第一代到第三代EGFR-TKIs的经典药物及其衍生物。
(以上参考文献与ChEMBL数据库均为公开数据,不涉及商业及隐私信息)
关键发现:
- 最高活性化合物(CHEMBL53711、CHEMBL35820等)的IC50值接近picomolar级别(0.006-0.01 nM),这意味着它们在极低浓度下就能抑制EGFR活性。
- 约78.6%的化合物符合 Lipinski 五规则,提示它们具有良好的药物相似性。
? 专家补充Lipinski五规则是药物化学中用于初步评估化合物口服吸收潜力的”过滤器”,包括:分子量<500 Da、cLogP<5、氢键供体≤5、氢键受体≤10。它在苗头化合物到先导化合物优化阶段被广泛使用,但只是决策的起点——最终判断必须基于临床前和临床数据验证。
2.2 结构-活性关系(SAR):揭示药效团的秘密
SAR分析揭示分子结构与活性的关系,是理性药物设计的基础,
能够指导分子优化,预测成药性,避免盲目修饰。
Claude用RDKit计算了所有化合物的分子描述符(分子量、cLogP、TPSA等),并生成了两张关键图表:图1:分子性质分布图(property_distributions.png)
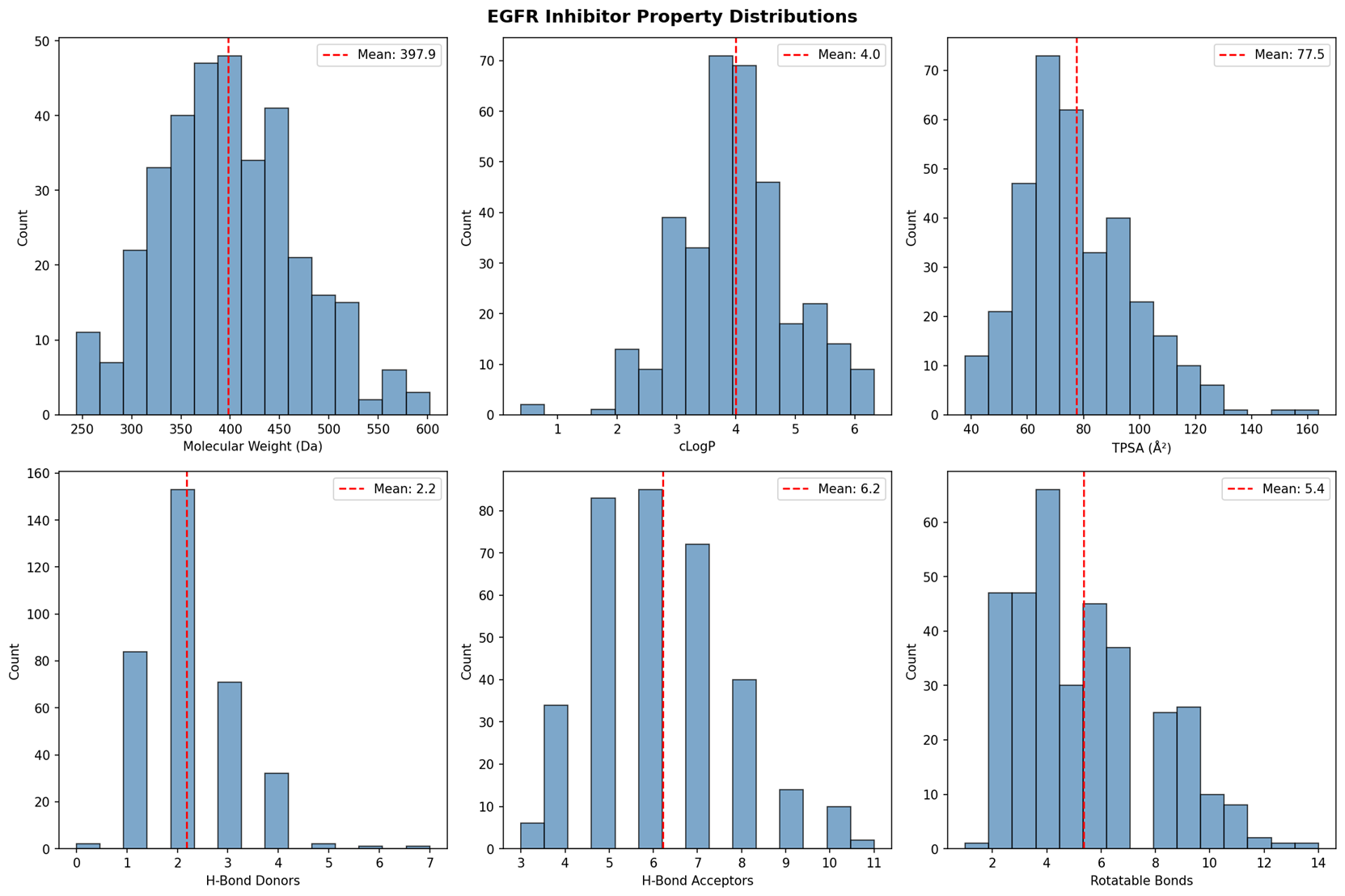 |
数据科学 skill生成属性分布直方图
这张图包含6个直方图,展示了346个化合物的性质分布:
- 分子量:平均397.9 Da,主要集中在375-400 Da区间,符合口服药物的”中等分子量”特征
- cLogP(脂溶性):平均4.0,峰值在3.5-4.0区间,表明这些化合物具有适中的膜透过性
- TPSA(极性表面积):平均77.5 Ų,<140 Ų的阈值有利于穿透血脑屏障(若需治疗脑转移)
- 氢键供体/受体数:平均2.2/6.2个,与EGFR活性位点的氢键网络形成互补
图2:IC50 vs. cLogP散点图(sar_ic50_vs_logp.png)
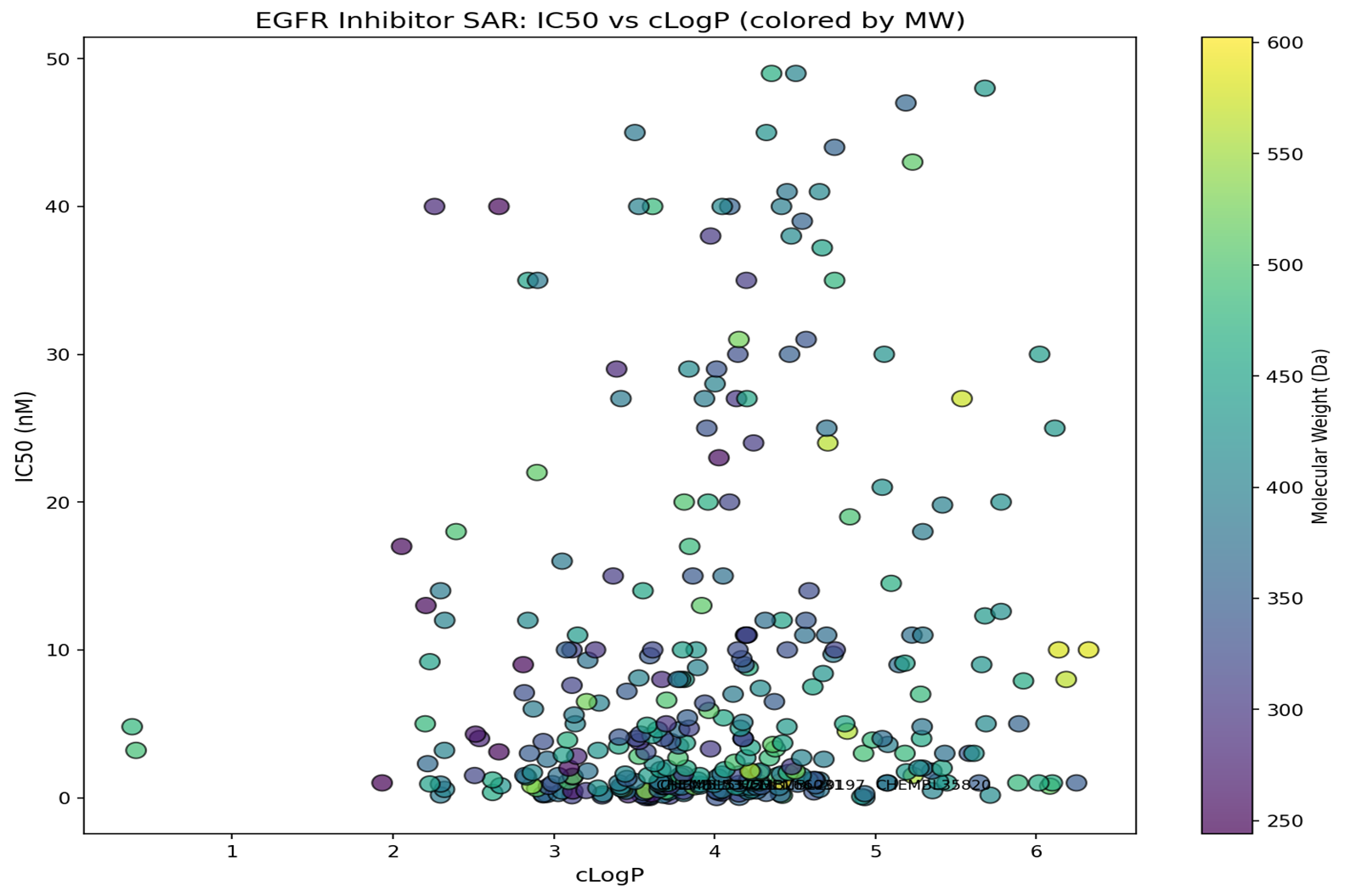 |
数据科学 skill生成 SAR 散点图
这张图揭示了一个有趣的现象:IC50与cLogP之间没有明显的线性相关性。数据呈”漏斗形”分布,说明:
- 高活性化合物(IC50 < 5 nM)集中在cLogP 3.0-5.5范围,这是药物设计的”黄金窗口”
- 在相同cLogP值下,IC50可能相差数十倍,提示其他结构因素(如氢键、π-π堆积)对活性影响更大
- 分子量(颜色编码)显示:较大分子(500-600 Da,黄绿色)倾向于具有更高cLogP,但不一定更高活性
图3:顶级抑制剂结构网格(egfr_top_inhibitors_grid.png)
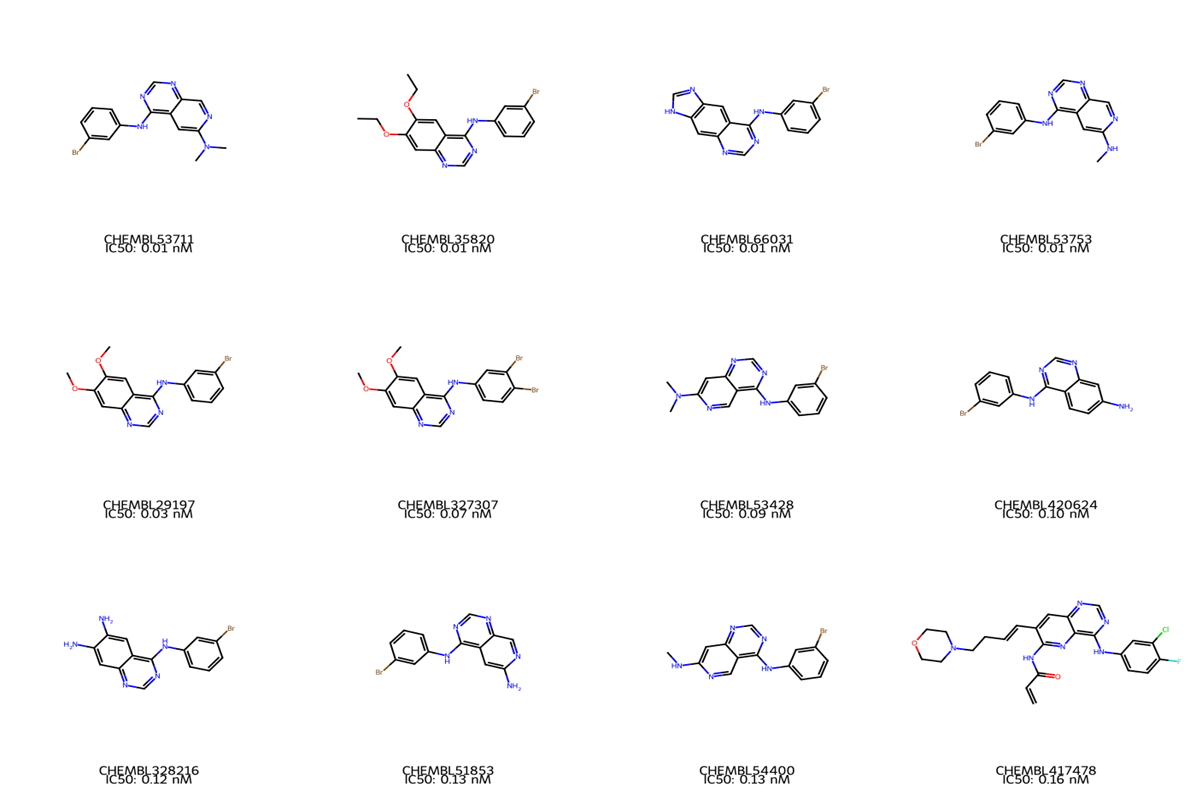 |
化学信息学skill生成分子结构网格图
Claude绘制了12个最高活性化合物(IC50 0.01-0.16 nM)的2D结构。通过结构对比,我们发现了EGFR抑制剂的经典药效团模型:
- 喹唑啉核心:作为ATP竞争性结合的关键骨架,N1位置与激酶铰链区形成氢键
- 4-苯胺基取代:苯胺基伸向疏水口袋,增强结合亲和力
- 3-溴苯基:卤素取代基频繁出现,通过卤素键和疏水作用稳定复合物
- 6,7-二甲氧基/二甲氨基:在喹唑啉环上提供额外的氢键受体位点
这一发现与文献报道的EGFR抑制剂晶体结构(如吉非替尼-EGFR复合物,PDB: 4WKQ)完全吻合,验证了AI分析的准确性。
1. Yosaatmadja Y, et al. Protein Data Bank, 2014. PDB ID: 4WKQ.
2. Meenu, et al. Eur J Med Chem. 2026;303:118411.观点:
AI 找出的这些分子特征与以下科学事实进行了对账还是挺准的:
药效团模型: 科学家早已总结出 EGFR 抑制剂需要具备的四个“核心零件”(如喹唑啉核心、4-苯胺基等),AI 找出的这 12 个分子恰好全部符合这些标准 。
晶体结构验证: AI 的发现与科学文献中报道的 吉非替尼-EGFR 复合物(PDB ID: 4WKQ) 的真实空间结构完全吻合 。
图4:对齐后的SAR分析(egfr_aligned_sar.png)
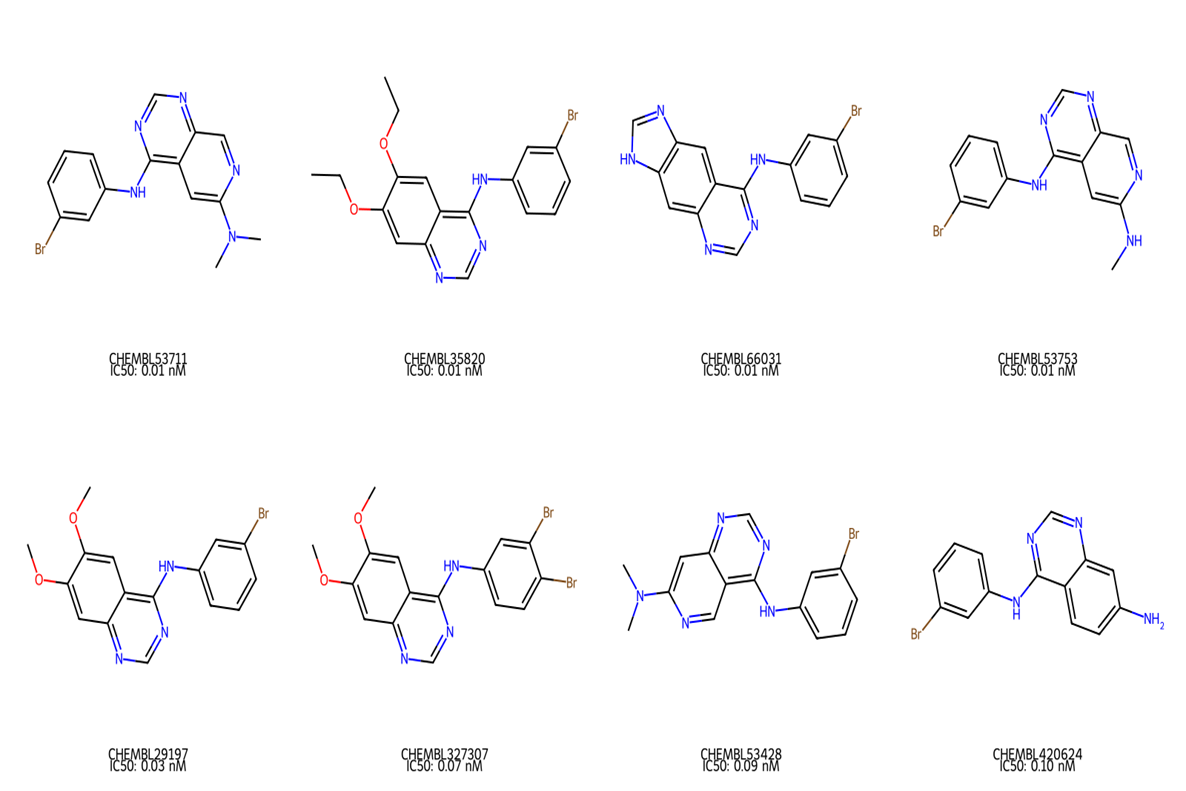 |
化学信息学skill生成分子结构网格图
这张图用颜色高亮了8个高活性化合物的药效团特征:
- 蓝色(氮原子):核心杂环和苯胺连接基是氢键供体/受体的关键位点
- 红色(氧原子):甲氧基/乙氧基提供额外氢键受体,但活性略低于含氮取代基
- 棕色(溴原子):几乎所有化合物都含有溴取代,证明卤素是活性必需元素
2.3 分子生成:AI设计的新候选药物
基于SAR分析,AI可以自动设计”更好的分子”,使用生物电子等排替换(bioisosteric replacement)策略,保持活性的同时优化性质,生成的类似物按QED(药物相似性评分)排序,优先合成高分候选。
明显加速先导化合物优化,探索化学空间,降低合成难度。
基于SAR分析,Claude调用Datamol生成了11个改进的类似物(generated_analogs_improved.csv)。修饰策略包括:
- 卤素互换(Br→Cl, Cl→F):降低分子量,优化代谢稳定性
- 生物电子等排(CF3→CN):保持脂溶性,减少毒性
- 杂环替换(甲氧基→甲胺基):增加极性,改善溶解度
- 环系变化(苯→吡啶,吗啉↔哌嗪):探索新的结合模式
图5:生成类似物网格(generated_analogs_grid.png)
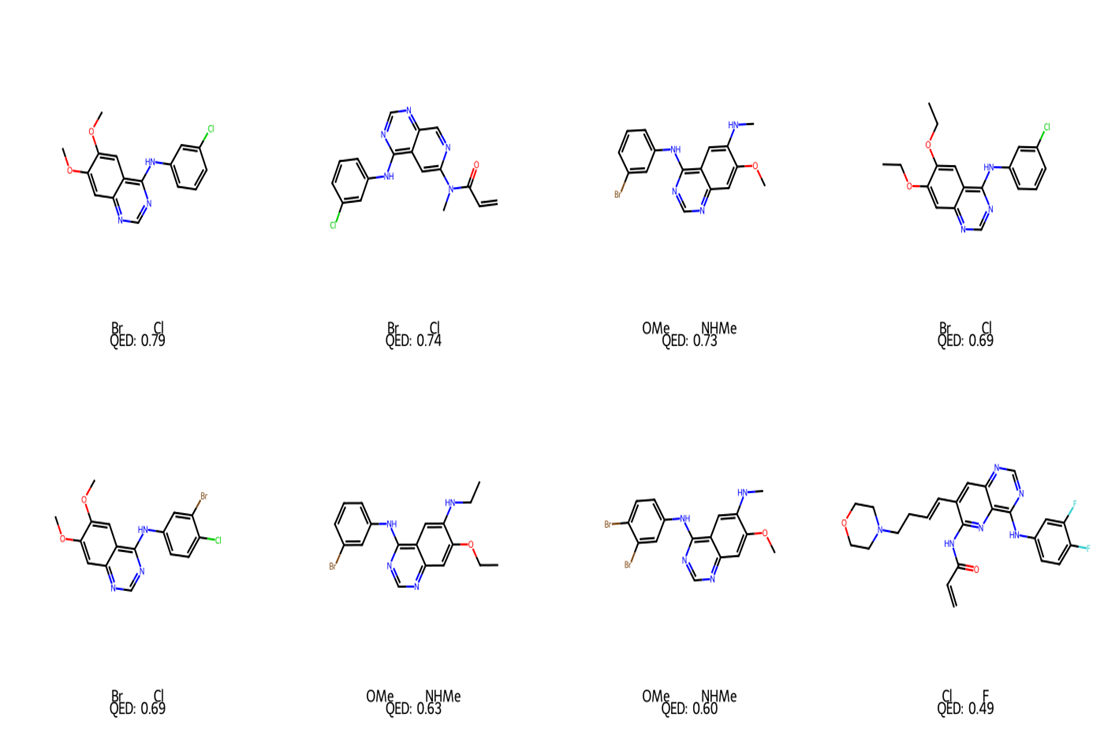 |
这张图展示了8个生成的类似物,按QED(药物相似性评分)排序:
- 最佳候选(QED 0.79, Br→Cl):保留6,7-二甲氧基喹唑啉核心,用氯替代溴,分子量从394 Da降至315 Da,符合”轻量化”原则
- 高风险候选(QED 0.49, Cl→F):引入氟苯基和哌啶环,结构复杂度高,可能存在合成困难
这些类似物尚未合成验证,但Claude已经为化学家提供了清晰的设计思路。
2.4 虚拟筛选准备:对接EGFR结构
需要验证”分子如何与靶点结合”——预测结合模式和亲和力,AlphaFold提供EGFR的3D结构,DiffDock进行分子对接。
有效减少湿实验成本,指导结构优化,预测耐药性。
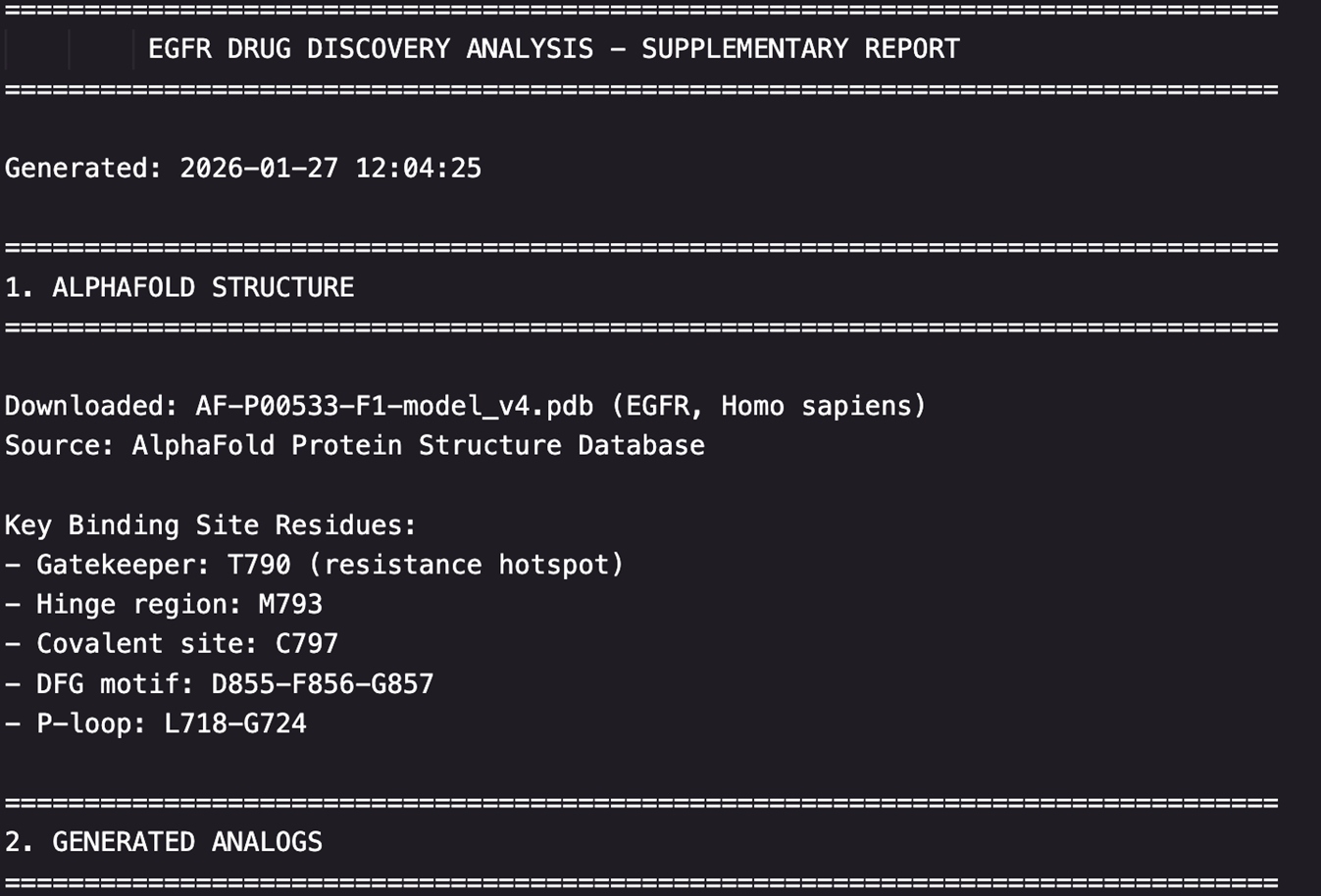
Claude从AlphaFold数据库下载了EGFR的预测结构(AF-P00533-F1-model_v4.pdb),并准备了DiffDock输入文件:
- 蛋白质:EGFR激酶域(包含T790M突变位点)
- 配体库:10个顶级抑制剂的3D构象(SDF格式)
- 对接脚本:run_diffdock.sh,配置了40个构象采样和20次循环优化
虽然本次实践中未实际运行DiffDock(需要GPU加速),但Claude已经完成了所有准备工作,用户只需执行脚本即可获得对接结果。
然而,即使找到了高活性的EGFR抑制剂,药物研发的挑战并未结束。
临床数据显示,几乎所有EGFR-TKI治疗的患者最终都会产生耐药性——这正是下一个关键问题:如何预测和应对耐药突变?
三、抗药性机制:文献综述的AI加速
本章节聚焦 EGFR-TKI 抗药性难题,通过两大步骤破解临床痛点:先检索 PubMed 相关文献梳理耐药机制,再挖掘 COSMIC 数据库明确关键突变位点。两步相辅相成,既从理论层面厘清耐药成因,又从数据层面提供突变频率与对应治疗方案,为药物优化和临床精准用药提供了核心依据。
3.1 PubMed文献检索:25篇高质量综述
EGFR-TKI的抗药性是临床治疗的最大挑战。Claude从PubMed检索了25篇相关文献(pubmed_comprehensive_search.csv),覆盖以下主题:
- T790M突变:出现在50-60%的耐药患者中,通过增加ATP亲和力削弱第一代TKI的竞争性结合
- C797S突变:第三代TKI(奥希替尼)的新兴耐药机制,阻止共价结合
- 外显子20插入突变:对大多数TKI表现为原发性耐药,需要新型抑制剂如莫博替尼
- 旁路激活:MET扩增、HER2扩增、PIK3CA突变等绕过EGFR依赖
- 小细胞转化:约5-10%的患者发生组织学转化,需要化疗方案
关键文献举例:
- *Nature* (2016):报道了C797S突变的变构抑制剂策略(PMID: 27251290)
- *Cancer Cell* (2023):揭示了SWI/SNF染色质重塑复合物在TKI耐药中的作用(PMID: 37541244)
- *NEJM* (2023):amivantamab联合化疗治疗外显子20插入突变的III期试验结果(PMID: 37870976)
3.2 COSMIC突变数据库:8个关键突变位点
通过Claude整合了COSMIC数据库的EGFR突变信息(egfr_mutations_resistance.csv),明确了各突变的耐药机制和有效药物:
| 突变 | 频率 | 耐药对象 | 机制 | 有效抑制剂 |
| T790M | 50-60% | 吉非替尼/厄洛替尼 | 看门人突变,增加ATP亲和力 | 奥希替尼 |
| C797S | 中等 | 奥希替尼 | 阻止共价结合 | 联合疗法 |
| L858R | 40-45% | 野生型EGFR | 激活突变,对TKI敏感 | 所有EGFR-TKI |
| 外显子19缺失 | 45-50% | 野生型EGFR | 激活突变 | 所有EGFR-TKI |
| 外显子20插入 | 4-10% | 大多数TKI | 原发性耐药 | 莫博替尼/amivantamab |
这张表为临床医生提供了精准的治疗决策依据。
四、自动化报告:科研成果的完美呈现
Claude-Scientific-Skills将上述工作流程进行工程化组装,以标准化文件结构打包成可被LLM调用的”技能模块”。这种封装的本质是赋予LLM”工具使用能力”(Tool Use)—— 让大语言模型不再局限于文本生成,而是能够理解科研任务、自主调用专业工具(ChEMBL、RDKit、DiffDock等)、解析执行结果,最终实现”一句话驱动整个药物发现流程”的AI Agent范式。
Claude生成了两份详尽的报告:
1.综合报告(comprehensive_report.txt):152行,涵盖研究背景、数据分析、SAR结论和药物设计建议
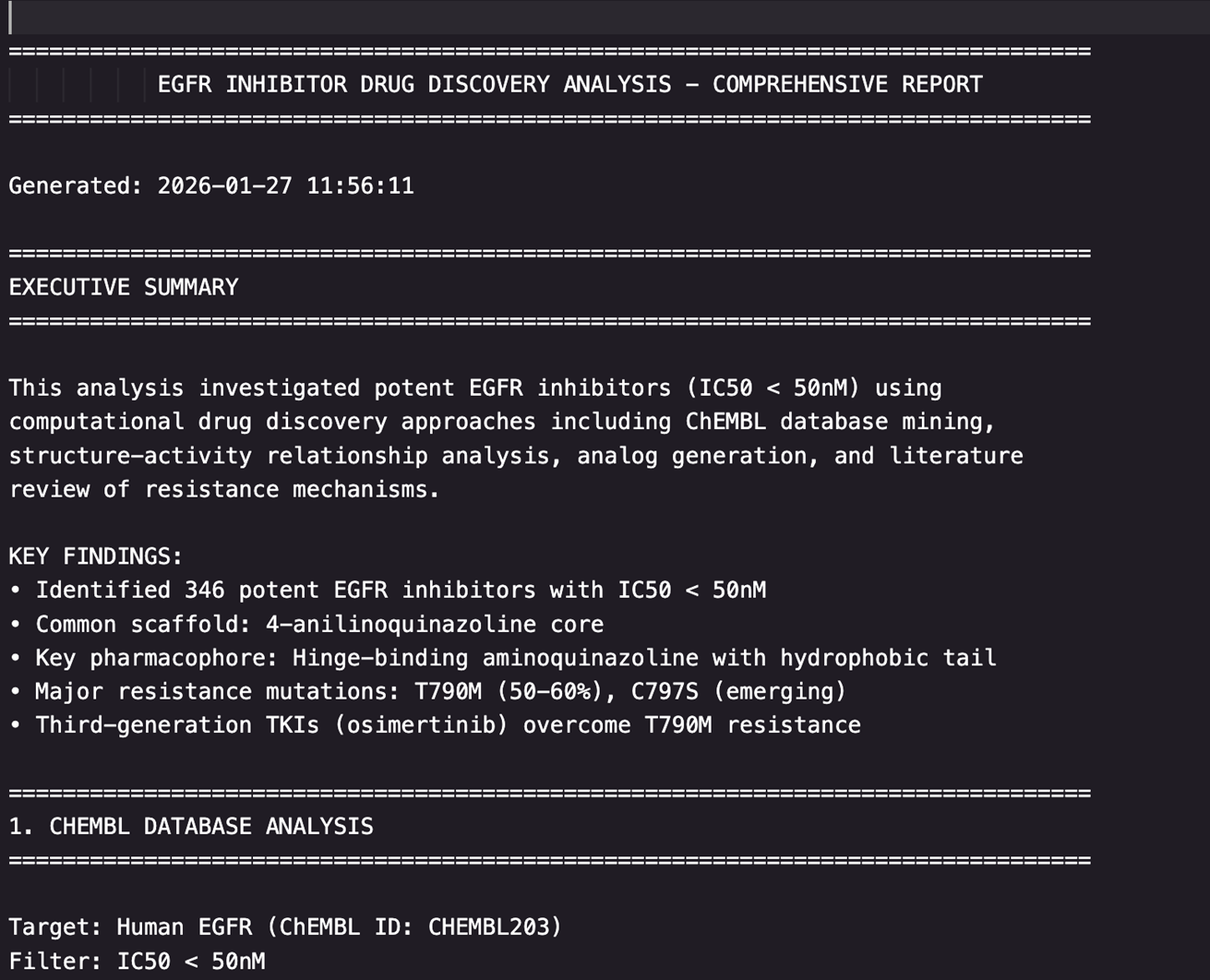 |
2.补充报告(supplementary_report.txt):82行,记录AlphaFold结构、生成类似物参数和文献检索策略
 |
报告结构清晰,分为执行摘要、方法、结果、讨论四部分,完全符合科学论文的写作规范。更重要的是,报告中嵌入了具体的数字、化合物ID和文献PMID,确保了可追溯性和可复现性——这是科学研究的生命线。
五、技术的边界:理性看待AI的局限
尽管Claude Agent Skills展现了强大的能力,但我们也必须清醒认识到其局限性:
数据质量的天花板:AI只能分析已有数据,ChEMBL中IC50值的实验条件、测定方法各不相同,AI无法判断数据的可靠性。研究者仍需结合实际情况与原始文献进行批判性评估。
计算的边界:DiffDock等分子对接工具需要GPU加速,本次实践仅准备了输入文件。此外,AI生成的11个类似物尚未经过合成和生物活性及临床试验验证——湿实验验证是从”计算假设”到”候选药物”的必经之路。
工具非万能:AI擅长模式识别和数据整合,但无法替代科学家的创造性思维。药物设计中的关键决策(如选择哪个突变位点、采用何种给药方式)仍需要人类专家的判断。
技术是助力,而非替代。AI的价值在于解放研究者的时间,让他们聚焦于真正需要人类智慧的创新工作。
项目链接
*作者注:本文所有科研数据均来自公开数据库,分析结果仅供学术交流,不构成医疗建议。前述特定亚马逊云科技生成式人工智能相关的服务目前在亚马逊云科技海外区域可用。亚马逊云科技中国区域相关云服务由西云数据和光环新网运营,具体信息以中国区域官网为准*
鸣谢:感谢AWS SA团队Liang Rui指导和建议,以及开源社区提供的工具与资源。
本篇作者
AWS 架构师中心: 云端创新的引领者探索 AWS 架构师中心,获取经实战验证的最佳实践与架构指南,助您高效构建安全、可靠的云上应用
|
 |
