AWS Compute Blog
Building High-Throughput Genomics Batch Workflows on AWS: Introduction (Part 1 of 4)
This post is courtesy of Aaron Friedman – Healthcare and Life Sciences Partner Solutions Architect at AWS and Angel Pizarro – Scientific Computing Technical Business Development Manager at AWS
Deriving insights from data is foundational to nearly every organization, and many customers process high volumes of data every day. One common requirement of customers in life sciences is the need to analyze these data in a high-throughput fashion without sacrificing time-to-insight.
Such analyses, which tend to be composed of a series of massively parallel processes (MPP) are well suited to the AWS Cloud. Many AWS customers and partners today, such as Illumina, DNAnexus, Seven Bridges Genomics, AstraZeneca, UCSC Genomics Institute, and Human Longevity, Inc., have built scalable and elastic genomics processing solutions on AWS.
One such use case is genomic sequencing. Modern DNA sequencers, such as Illumina’s NovaSeq, can produce multiple terabytes of raw data each day. The data must then be processed into meaningful information that clinicians and research can act on in a timely fashion. This processing of genomic data is commonly referred to as “secondary analysis”.
Most common secondary analysis workflows take raw reads generated from sequencers and then process them in a multi-step workflow to identify the variation in a biological sample compared to a standard genome reference. The individual steps are normally similar to the following:
- DNA sequences are mapped to a standard genome reference by use of an alignment algorithm, such as Smith-Waterman or Burrows-Wheeler.
- After the sequences are mapped to the reference, the differences are identified as single nucleotide variations, insertions, deletions, or complex structural variation in a process known as variant calling.
- The resulting variants are often combined with other information to identify genomic variants highly correlated with disease or drug response. They might also be analyzed in the context of clinical data such as to identify disease susceptibility or state for a patient.
- Along the way, quality metrics are collected or computed to ensure that the generated data is of the appropriate quality for use in requisite research or clinical settings.
In this post series, you can build a secondary analysis workflow similar to the one just described. Here is a diagram of the workflow:
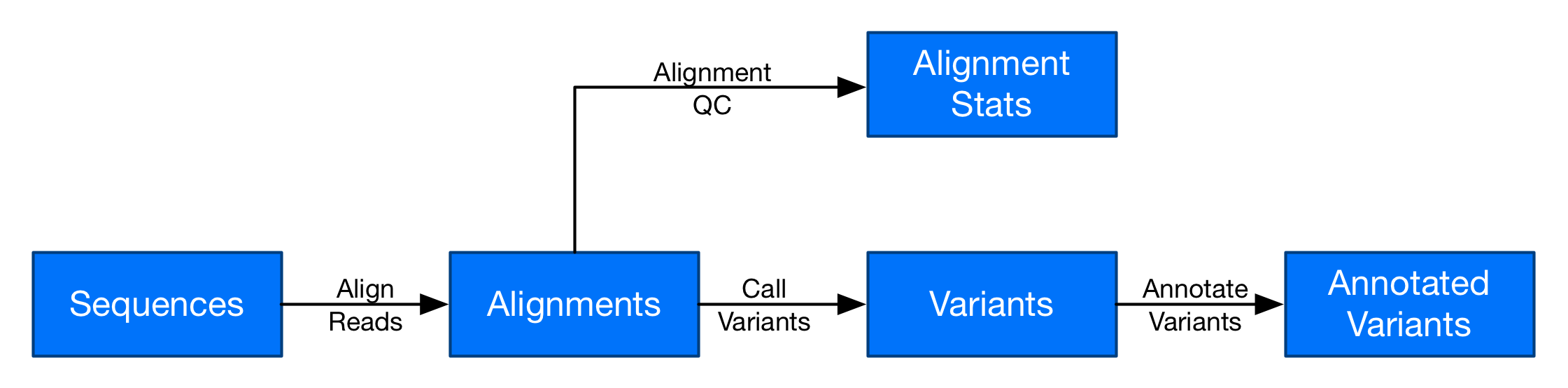
Secondary analysis is a batch workflow
At its core, a genomics pipeline is similar to a series of Extract Transform and Load (ETL) steps that convert raw files from a DNA sequencer to a list of variants for one or more individuals. Each step extracts a set of input files from a data source, processes them as a compute-intensive workload (transform), and then loads the output into another location for subsequent storage or analysis.
These steps are often chained together to build a flexible genomics processing workflow. The files can then be used for downstream analysis, such as population scale analytics with Amazon Athena or Spark on Amazon EMR. These ETL processes can be represented as individual batch steps in an overall workflow.
When we discuss batch processing with customers, we often focus on the following three layers:
Jobs (analytical modules): These jobs are individual units of work that take a set of inputs, run a single process, and produce a set of outputs. In this series, you use Docker containers to define these analytical modules. For genomics, these commonly include alignment, variant calling, quality control, or another module in your workflow. Amazon ECS is an AWS service that orchestrates and runs these Docker containerized modules on top of Amazon EC2 instances.
Batch engine: This is a framework for submitting individual analytical modules with the appropriate requirements for computational resources, such as memory and number of CPUs. Each step of the analysis pipeline requires a definition of how to run a job:
- Computational resources (disk, memory, CPU)
- The compute environment to run it in (Docker container, runtime parameters)
- Information about the priority of different jobs
- Any dependencies between jobs
You can leverage concepts such as container placement and bin packing to maximize performance of your genomic pipeline while concurrently optimizing for cost. We will use AWS Batch for this layer. AWS Batch dynamically provisions the optimal quantity and type of compute resources (for example, CPU or memory optimized instances) based on the volume and specific resource requirements of the submitted batch jobs.
Workflow orchestration: This layer sits on top of the batch engine and allows you to decouple your workflow definition from the execution of the individual jobs. You can envision this layer as a state machine where you define a series of steps and pass appropriate metadata between states. We will use AWS Lambda to submit the jobs to AWS Batch and use AWS Step Functions to define and orchestrate the workflow.
Architecture
In the next three posts, you build a genome analysis pipeline using the following architecture. You don’t explicitly build the grayed out section, but we wanted to include them in the diagram as they are natural extensions to the core architecture. We discuss these and other extensions, briefly, in the concluding post. All code related to this blog series can be found in the associated GitHub repository here.
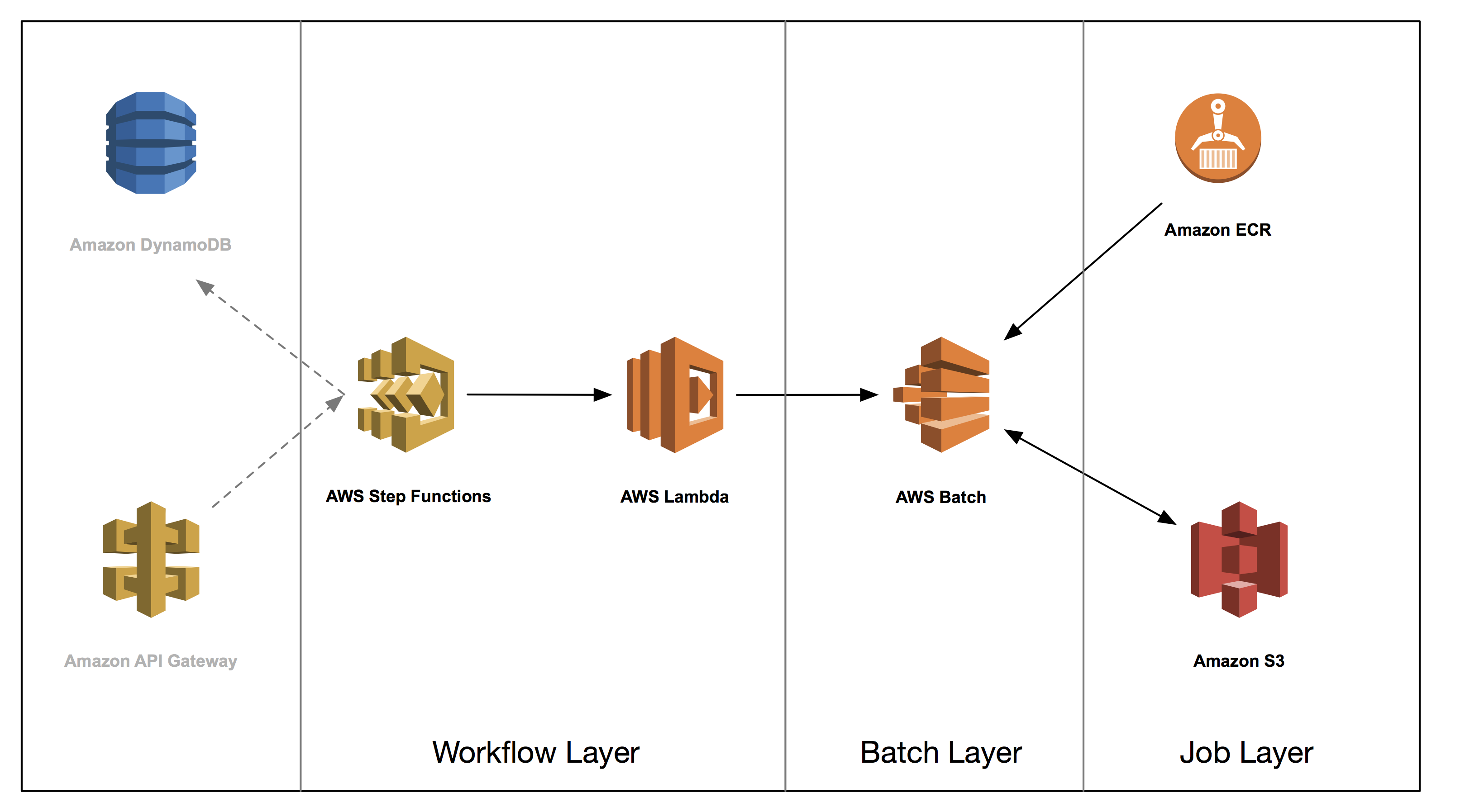
Part 2 covers the job layer. We demonstrate how you can package bioinformatics applications in Docker containers, and discuss best practices when developing these containers for use in a multitenant batch environment.
Part 3 dives deep into the batch, or data processing layer. We discuss common considerations for deploying Docker containers to be used in batch analysis as well as demonstrate how you can use AWS Batch for a scalable and elastic batch engine.
Part 4 dives into workflow layer orchestration. We show how you might architect that layer with AWS services. You take the components built in parts 2 and 3 and combine them into an entire secondary analysis workflow. This workflow manages dependencies as well as continually checking the progress of existing jobs. We conclude by running a secondary analysis end-to-end for under $1 and discuss some extensions you can build on top of this core workflow.
What you can expect to learn
At the end of this series, you will have built a scalable and elastic solution to process genomes on AWS, as well as gained a general understanding of architectural choices available to you. The solution is generally applicable to other workloads, such as image processing. Even non-life science workloads such as trade analytics in financial services can benefit.
In your solution, you use Amazon EC2 Spot Instances to optimize for cost. Spot Instances allow you to bid on spare EC2 compute capacity, which can save up to 90% off of traditional On-Demand prices. In many cases, this translates into the ability to analyze genomes at scale for as low as $1 per analysis.
Let’s get building!
Other posts in this four-part series:
Please leave any questions and comments below.