AWS Compute Blog
Building High-Throughput Genomics Batch Workflows on AWS: Job Layer (Part 2 of 4)
This post is courtesy of Aaron Friedman – Healthcare and Life Sciences Partner Solutions Architect at AWS and Angel Pizarro – Scientific Computing Technical Business Development Manager at AWS
This post is the second in a series on how to build a genomics workflow on AWS. In Part 1, we introduced a general architecture, shown below, and highlighted the three common layers in a batch workflow:
- Job
- Batch
- Workflow
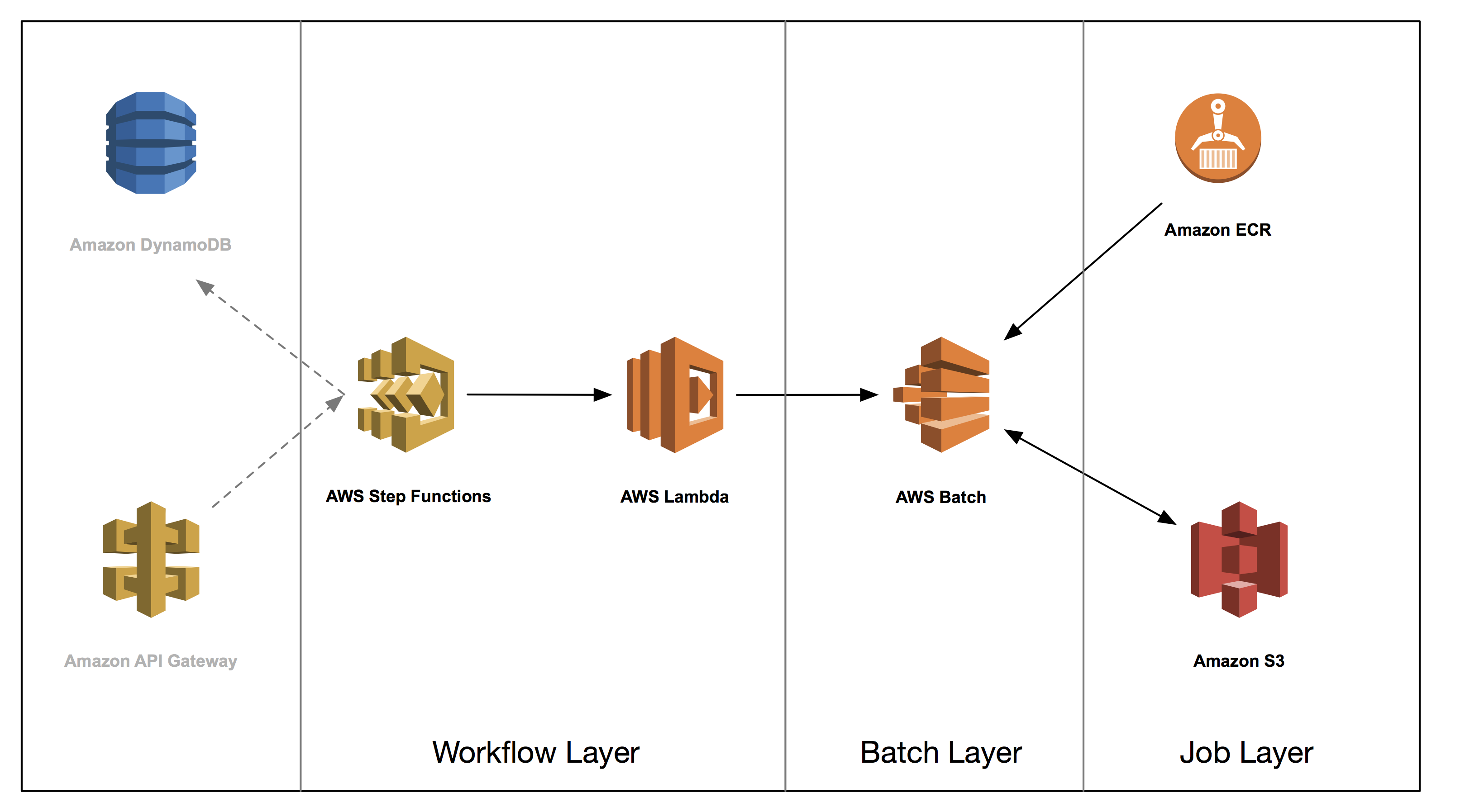
In Part 2, you tackle the job layer and package a set of bioinformatics applications into Docker containers and store them in Amazon ECR. We illustrate some common patterns and best practices for these containers, such as how you can effectively use Amazon S3 to exchange data between jobs.
ECR is a fully managed Docker container registry that makes it easy for developers to store, manage, and deploy Docker container images. ECR is integrated with Amazon ECS, simplifying your development to a production workflow. ECR eliminates the need to operate your own container repositories or worry about scaling the underlying infrastructure by hosting your images in a highly available and scalable architecture. You can integrate with IAM to provide resource-level control for each repository.
All code related to this blog series can be found in the associated GitHub repository here.
Packaging an application in a Docker container
Genomic analysis often relies on open source software that is developed by academic groups or open-sourced by industry leaders. These applications have a range of requirements for libraries and reference data, and are typically executed using a Linux command line interface.
Docker containers provide a consistent, reproducible run-time environment for applications to run in, which results in reproducible results. Consequently, containerization of the applications using Docker has received much attention from the bioinformatics community, resulting in the development of application registries such as Dockstore.org, BioContainers, and the Galaxy Tool Shed. In this post, we cover several good practices for packing genomics applications in Docker containers, including:
- Building your Dockerfile
- Dealing with job multitenancy
- Sharing data between jobs
Building your Dockerfile
Your Dockerfile contains all of the commands that you use to package your Docker container. In it, you pick a base image to build from, include any metadata to attribute to the image, describe how to build and configure the environment, and how to access the code running within it.
We recommend that you adopt a standard set of conventions for your Dockerfiles. Add metadata to describe the contained application, and have an order set of sections for application packaging needs. Here is an example from the provided Isaac Dockerfile:
FROM python:2.7
# Metadata
LABEL container.base.image="python:2.7"
LABEL software.name="iSAAC"
LABEL software.version="03.17.03.01"
LABEL software.description="Aligner for sequencing data"
LABEL software.website="https://github.com/Illumina/Isaac3"
LABEL software.documentation="https://github.com/Illumina/Isaac3/blob/master/src/markdown/manual.md"
LABEL software.license="GPLv3"
LABEL tags="Genomics"
RUN apt-get -y update && \
apt-get -y install zlib1g-dev gnuplot && \
apt-get clean
RUN pip install boto3 awscli
RUN git clone https://github.com/Illumina/Isaac3.git && cd /Isaac3 && git checkout 6f0191a4e0d4b332e8f34b7ced57dc6e6eb4f2f1
RUN mkdir /iSAAC-build /isaac
WORKDIR /iSAAC-build
RUN /Isaac3/src/configure --prefix=/isaac
RUN make
RUN make install
WORKDIR /
RUN rm -rf /Isaac3 /iSAAC-build
RUN chmod -R +x /isaac/bin/
ENV PATH="/isaac/bin:$PATH"
ENV LD_LIBRARY_PATH="/usr/local/lib:/usr/lib:/isaac/libexec:${LD_LIBRARY_PATH}"
COPY isaac/src/run_isaac.py /
COPY common_utils /common_utils
ENTRYPOINT ["python", "/run_isaac.py"]There are many ways to architect Docker containers, but we wanted to consolidate some recommendations that we have observed our customers successfully using:
- Work off of a base image that satisfies most dependencies across a set of applications. In the code provided, we often use the official
python:2.7image from Docker Hub. - In the above Dockerfile, you can see that we have separated out the metadata, underlying system and library dependencies, application-specific dependencies, and the installation requirements into a logical ordered set for easier maintenance. It’s worth noting that if you are building a production application, you would traditionally have a golden set of images to build from and application artifacts to install that have been validated with internal processes.
- Instead of building large datasets into the container itself, we recommend that you download reference data at runtime instead. This allows the decoupling of the algorithm from reference data updates, which could happen nightly. It also allows you to download a subset of all reference data for an algorithm that is specific to the running analysis, for example the particular species under study.
- Provide an application entry point to both expose and limit the default functionality of the container image. In this example, we created a simple Python script that takes care of downloading the dependencies from S3, such as reference data for your analysis, on the fly from a set of provided runtime parameters and stage results back into S3. We dive deeper into this script in the following section.
Often these shared dependencies are a mix of packaged code that is easily installable (in this case via pip) or private modules usually shared as part of internal source repositories, as is the case here.
When you build the Docker images, you should take care to include both the necessary private modules within the context of the build. The example below shows how you would accomplish that given a directory context with some dependencies a few levels higher that the Dockerfile. In the project, we provided a Makefile for taking care of some of these items, but for clarity’s sake we issue the necessary Docker commands.
# Given the following partial directory tree structure
# .
# └── tools
# ├── common\_utils
# │ ├── __init__.py
# │ ├── job_utils.py
# │ └── s3_utils.py
# └── isaac
# └── docker
# ├── Dockerfile
# └── Makefile
# cd <git repository>/tools/isaac/docker
$ docker build -t isaac:03.17.03.01 \
-t isaac:latest \
-f Dockerfile ../..Job multitenancy and sharing data between jobs
Many bioinformatics tools have been developed to run in any Linux environment, and not necessarily optimized for cloud computing or multitenancy. To overcome these challenges, you can use a simple Python wrapper script for each tool that facilitates the deployment of a job.
Our tools have several of the same requirements, such as the need to read and write from S3 and deal with job multitenancy. For these common utilities, we built a separate common_utils package to import during the creation of the Docker image. These utilities deal with the previously mentioned common requirements, such as:
-
- Container placement
To make your workflow as flexible as possible, each job should run independently. As a result, you cannot necessarily guarantee that different jobs in the same overall workflow run on the same instance. Using S3 as the location to exchange data between containers enables you to decouple storage of your intermediate files from compute. The tools/common_utils/s3_utils.py script contains the functions required to leverage S3.
-
- Multitenancy
Multiple container jobs may run concurrently on the same instance. In these situations, it’s essential that your job writes to a unique subdirectory. An easy way to do this is to create a subfolder using a UUID and have your application write all of your data there.
-
- Cleanup
As your jobs complete and write the output back to S3, you can delete the scratch data on your instance generated by that job. This allows you to optimize for cost by reusing EC2 instances if there are jobs remaining in the queue, rather than terminating the EC2 instances. As you ensure that you’re writing to a unique subdirectory in the multitenancy solution, you can simply delete that subdirectory to minimize your storage footprint.
Each of the Python wrappers takes in all of the requisite data dependencies, residing in S3, as command-line arguments and any other necessary commands to run the tool it wraps. It then handles all of the file downloading, running the bioinformatics tool, and uploading the files back to S3. For more information about each of these tools, see the READMEs for each individual tool.
Deploying images to Amazon ECR
Next, publish the Docker images to ECR. The first example below creates an ECR repository and collects the URI you provide to Docker in order to push the image to ECR. If a repository already exists for the container image, query for it, as shown in the second example.
# Create an ECR repository for Isaac, then copy the `repositoryUri` into a variable
$ REPO\_URI=$(aws ecr create-repository \
--repository-name isaac \
--output text --query "repository.repositoryUri")
# If the repository already exists, then
# query ECR for the `repositoryUri`
$ REPO\_URI=$(aws ecr describe-repositories \
--repository-names isaac \
--output text --query "repositories[0].repositoryUri")After you have a repository URI, you can tag the container image and push it to ECR.
$ eval $(aws ecr get-login)
$ docker tag isaac:latest $(REPO\_URI):latest
$ docker push $(REPO\_URI):latest
$ docker tag isaac:03.17.03.01 $(REPO\_URI):03.17.03.01
$ docker push $(REPO\_URI):03.17.03.01Conclusion
In part 2 of this series, we showed a practical example of packaging up a bioinformatics application within a Docker container, and publishing the container image to an ECR repository. Along the way, we discussed some generally applicable best practices and design decisions specific to this demonstration project.
Now that you have built one of the Docker containers, you can go ahead and build each of the containers. We have provided all of the necessary code within a GitHub repository and within that repository are specific instructions, as well as some helpers for building container images using GNU make.
In Part 3 of this series, you’ll dive deep into the batch processing layer and how to leverage the packaged applications within AWS Batch.
Other posts in this four-part series:
Please leave any questions and comments below.