AWS Compute Blog
Building a tightly coupled molecular dynamics workflow with multi-node parallel jobs in AWS Batch
Contributed by Amr Ragab, HPC Application Consultant, AWS Professional Services and Aswin Damodar, Senior Software Development Engineer, AWS Batch
At Supercomputing 2018 in Dallas, TX, AWS announced AWS Batch support for running tightly coupled workloads in a multi-node parallel jobs environment. This AWS Batch feature enables applications that require strong scaling for efficient computational workloads.
Some of the more popular workloads that can take advantage of this feature enhancement include computational fluid dynamics (CFD) codes such as OpenFoam, Fluent, and ANSYS. Other workloads include molecular dynamics (MD) applications such as AMBER, GROMACS, NAMD.
Running tightly coupled, distributed, deep learning frameworks is also now possible on AWS Batch. Applications that can take advantage include TensorFlow, MXNet, Pytorch, and Chainer. Essentially, any application scaling that benefits from tightly coupled–based scalability can now be integrated into AWS Batch.
In this post, we show you how to build a workflow executing an MD simulation using GROMACS running on GPUs, using the p3 instance family.
AWS Batch overview
AWS Batch is a service providing managed planning, scheduling, and execution of containerized workloads on AWS. Purpose-built for scalable compute workloads, AWS Batch is ideal for high throughput, distributed computing jobs such as video and image encoding, loosely coupled numerical calculations, and multistep computational workflows.
If you are new to AWS Batch, consider gaining familiarity with the service by following the tutorial in the Creating a Simple “Fetch & Run” AWS Batch Job post.
Prerequisites
You need an AWS account to go through this walkthrough. Other prerequisites include:
- Launch an ECS instance, p3.2xlarge with a NVIDIA Tesla V100 backend. Use the Amazon Linux 2 AMIs for ECS.
- In the ECS instance, install the latest CUDA 10 stack, which provides the toolchain and compilation libraries as well as the NVIDIA driver.
- Install nvidia-docker2.
- In your /etc/docker/daemon.json file, ensure that the default-runtime value is set to nvidia.
{
"runtimes": {
"nvidia": {
"path": "nvidia-container-runtime",
"runtimeArgs": []
}
},
"default-runtime": "nvidia"
}
- Finally, save the EC2 instance as an AMI in your account. Copy the AMI ID, as you need it later in the post.
Deploying the workload
In a production environment, it’s important to efficiently execute the compute workload with multi-node parallel jobs. Most of the optimization is on the application layer and how efficiently the Message Passing Interface (MPI) ranks (MPI and OpenMP threads) are distributed across nodes. Application-level optimization is out of scope for this post, but should be considered when running in production.
One of the key requirements for running on AWS Batch is a Dockerized image with the application, libraries, scripts, and code. For multi-node parallel jobs, you need an MPI stack for the tightly coupled communication layer and a wrapper script for the MPI orchestration. The running child Docker containers need to pass container IP address information to the master node to fill out the MPI host file.
The undifferentiated heavy lifting that AWS Batch provides is the Docker-to-Docker communication across nodes using Amazon ECS task networking. With multi-node parallel jobs, the ECS container receives environmental variables from the backend, which can be used to establish which running container is the master and which is the child.
- AWS_BATCH_JOB_MAIN_NODE_INDEX—The designation of the master node in a multi-node parallel job. This is the main node in which the MPI job is launched.
- AWS_BATCH_JOB_MAIN_NODE_PRIVATE_IPV4_ADDRESS—The IPv4 address of the main node. This is presented in the environment for all children nodes.
- AWS_BATCH_JOB_NODE_INDEX—The designation of the node index.
- AWS_BATCH_JOB_NUM_NODES – The number of nodes launched as part of the node group for your multi-node parallel job.
If AWS_BATCH_JOB_MAIN_NODE_INDEX = AWS_BATCH_JOB_NODE_INDEX, then this is the main node. The following code block is an example MPI synchronization script that you can include as part of the CMD structure of the Docker container. Save the following code as mpi-run.sh.
#!/bin/bash
cd $JOB_DIR
PATH="$PATH:/opt/openmpi/bin/"
BASENAME="${0##*/}"
log () {
echo "${BASENAME} - ${1}"
}
HOST_FILE_PATH="/tmp/hostfile"
AWS_BATCH_EXIT_CODE_FILE="/tmp/batch-exit-code"
aws s3 cp $S3_INPUT $SCRATCH_DIR
tar -xvf $SCRATCH_DIR/*.tar.gz -C $SCRATCH_DIR
sleep 2
usage () {
if [ "${#@}" -ne 0 ]; then
log "* ${*}"
log
fi
cat <<ENDUSAGE
Usage:
export AWS_BATCH_JOB_NODE_INDEX=0
export AWS_BATCH_JOB_NUM_NODES=10
export AWS_BATCH_JOB_MAIN_NODE_INDEX=0
export AWS_BATCH_JOB_ID=string
./mpi-run.sh
ENDUSAGE
error_exit
}
# Standard function to print an error and exit with a failing return code
error_exit () {
log "${BASENAME} - ${1}" >&2
log "${2:-1}" > $AWS_BATCH_EXIT_CODE_FILE
kill $(cat /tmp/supervisord.pid)
}
# Set child by default switch to main if on main node container
NODE_TYPE="child"
if [ "${AWS_BATCH_JOB_MAIN_NODE_INDEX}" ==
"${AWS_BATCH_JOB_NODE_INDEX}" ]; then
log "Running synchronize as the main node"
NODE_TYPE="main"
fi
# wait for all nodes to report
wait_for_nodes () {
log "Running as master node"
touch $HOST_FILE_PATH
ip=$(/sbin/ip -o -4 addr list eth0 | awk '{print $4}' | cut -d/ -f1)
if [ -x "$(command -v nvidia-smi)" ] ; then
NUM_GPUS=$(ls -l /dev/nvidia[0-9] | wc -l)
availablecores=$NUM_GPUS
else
availablecores=$(nproc)
fi
log "master details -> $ip:$availablecores"
echo "$ip slots=$availablecores" >> $HOST_FILE_PATH
lines=$(sort $HOST_FILE_PATH|uniq|wc -l)
while [ "$AWS_BATCH_JOB_NUM_NODES" -gt "$lines" ]
do
log "$lines out of $AWS_BATCH_JOB_NUM_NODES nodes joined, check again in 1 second"
sleep 1
lines=$(sort $HOST_FILE_PATH|uniq|wc -l)
done
# Make the temporary file executable and run it with any given arguments
log "All nodes successfully joined"
# remove duplicates if there are any.
awk '!a[$0]++' $HOST_FILE_PATH > ${HOST_FILE_PATH}-
deduped
cat $HOST_FILE_PATH-deduped
log "executing main MPIRUN workflow"
cd $SCRATCH_DIR
. /opt/gromacs/bin/GMXRC
/opt/openmpi/bin/mpirun --mca btl_tcp_if_include eth0 \
-x PATH -x LD_LIBRARY_PATH -x
GROMACS_DIR -x GMXBIN -x GMXMAN -x GMXDATA \
--allow-run-as-root --machinefile
${HOST_FILE_PATH}-deduped \
$GMX_COMMAND
sleep 2
tar -czvf $JOB_DIR/batch_output_$AWS_BATCH_JOB_ID.tar.gz
$SCRATCH_DIR/*
aws s3 cp $JOB_DIR/batch_output_$AWS_BATCH_JOB_ID.tar.gz
$S3_OUTPUT
log "done! goodbye, writing exit code to
$AWS_BATCH_EXIT_CODE_FILE and shutting down my supervisord"
echo "0" > $AWS_BATCH_EXIT_CODE_FILE
kill $(cat /tmp/supervisord.pid)
exit 0
}
# Fetch and run a script
report_to_master () {
# get own ip and num cpus
#
ip=$(/sbin/ip -o -4 addr list eth0 | awk '{print $4}' | cut -d/ -f1)
if [ -x "$(command -v nvidia-smi)" ] ; then
NUM_GPUS=$(ls -l /dev/nvidia[0-9] | wc -l)
availablecores=$NUM_GPUS
else
availablecores=$(nproc)
fi
log "I am a child node -> $ip:$availablecores, reporting to the master node ->
${AWS_BATCH_JOB_MAIN_NODE_PRIVATE_IPV4_ADDRESS}"
until echo "$ip slots=$availablecores" | ssh
${AWS_BATCH_JOB_MAIN_NODE_PRIVATE_IPV4_ADDRESS} "cat >>
/$HOST_FILE_PATH"
do
echo "Sleeping 5 seconds and trying again"
done
log "done! goodbye"
exit 0
}
# Main - dispatch user request to appropriate function
log $NODE_TYPE
case $NODE_TYPE in
main)
wait_for_nodes "${@}"
;;
child)
report_to_master "${@}"
;;
*)
log $NODE_TYPE
usage "Could not determine node type. Expected (main/child)"
;;
esac
The synchronization script supports downloading the assets from Amazon S3 as well as preparing the MPI host file based on GPU scheduling for GROMACS.
Furthermore, the mpirun stanza is captured in this script. This script can be a template for several multi-node parallel job applications by just changing a few lines. These lines are essentially the GROMACS-specific steps:
. /opt/gromacs/bin/GMXRC
export OMP_NUM_THREADS=$OMP_THREADS
/opt/openmpi/bin/mpirun -np $MPI_THREADS --mca btl_tcp_if_include eth0 \
-x OMP_NUM_THREADS -x PATH -x LD_LIBRARY_PATH -x GROMACS_DIR -x GMXBIN -x GMXMAN -x GMXDATA \
--allow-run-as-root --machinefile ${HOST_FILE_PATH}-deduped \
$GMX_COMMAND
In your development environment for building Docker images, create a Dockerfile that prepares the software stack for running GROMACS. The key elements of the Dockerfile are:
- Set up a passwordless-ssh keygen.
- Download, and compile OpenMPI. In this Dockerfile, you are downloading the recently released OpenMPI 4.0.0 source and compiling on a NVIDIA Tesla V100 GPU-backed instance (p3.2xlarge).
- Download and compile GROMACS.
- Set up supervisor to run SSH at Docker container startup as well as processing the mpi-run.sh script as the CMD.
Save the following script as a Dockerfile:
FROM nvidia/cuda:latest
ENV USER root
# -------------------------------------------------------------------------------------
# install needed software -
# openssh
# mpi
# awscli
# supervisor
# -------------------------------------------------------------------------------------
RUN apt update
RUN DEBIAN_FRONTEND=noninteractive apt install -y iproute2 cmake openssh-server openssh-client python python-pip build-essential gfortran wget curl
RUN pip install supervisor awscli
RUN mkdir -p /var/run/sshd
ENV DEBIAN_FRONTEND noninteractive
ENV NOTVISIBLE "in users profile"
#####################################################
## SSH SETUP
RUN sed -i 's/PermitRootLogin prohibit-password/PermitRootLogin yes/' /etc/ssh/sshd_config
RUN sed 's@session\s*required\s*pam_loginuid.so@session optional pam_loginuid.so@g' -i /etc/pam.d/sshd
RUN echo "export VISIBLE=now" >> /etc/profile
RUN echo "${USER} ALL=(ALL) NOPASSWD:ALL" >> /etc/sudoers
ENV SSHDIR /root/.ssh
RUN mkdir -p ${SSHDIR}
RUN touch ${SSHDIR}/sshd_config
RUN ssh-keygen -t rsa -f ${SSHDIR}/ssh_host_rsa_key -N ''
RUN cp ${SSHDIR}/ssh_host_rsa_key.pub ${SSHDIR}/authorized_keys
RUN cp ${SSHDIR}/ssh_host_rsa_key ${SSHDIR}/id_rsa
RUN echo " IdentityFile ${SSHDIR}/id_rsa" >> /etc/ssh/ssh_config
RUN echo "Host *" >> /etc/ssh/ssh_config && echo " StrictHostKeyChecking no" >> /etc/ssh/ssh_config
RUN chmod -R 600 ${SSHDIR}/* && \
chown -R ${USER}:${USER} ${SSHDIR}/
# check if ssh agent is running or not, if not, run
RUN eval `ssh-agent -s` && ssh-add ${SSHDIR}/id_rsa
##################################################
## S3 OPTIMIZATION
RUN aws configure set default.s3.max_concurrent_requests 30
RUN aws configure set default.s3.max_queue_size 10000
RUN aws configure set default.s3.multipart_threshold 64MB
RUN aws configure set default.s3.multipart_chunksize 16MB
RUN aws configure set default.s3.max_bandwidth 4096MB/s
RUN aws configure set default.s3.addressing_style path
##################################################
## CUDA MPI
RUN wget -O /tmp/openmpi.tar.gz https://download.open-mpi.org/release/open-mpi/v4.0/openmpi-4.0.0.tar.gz && \
tar -xvf /tmp/openmpi.tar.gz -C /tmp
RUN cd /tmp/openmpi* && ./configure --prefix=/opt/openmpi --with-cuda --enable-mpirun-prefix-by-default && \
make -j $(nproc) && make install
RUN echo "export PATH=$PATH:/opt/openmpi/bin" >> /etc/profile
RUN echo "export LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/opt/openmpi/lib:/usr/local/cuda/include:/usr/local/cuda/lib64" >> /etc/profile
###################################################
## GROMACS 2018 INSTALL
ENV PATH $PATH:/opt/openmpi/bin
ENV LD_LIBRARY_PATH $LD_LIBRARY_PATH:/opt/openmpi/lib:/usr/local/cuda/include:/usr/local/cuda/lib64
RUN wget -O /tmp/gromacs.tar.gz http://ftp.gromacs.org/pub/gromacs/gromacs-2018.4.tar.gz && \
tar -xvf /tmp/gromacs.tar.gz -C /tmp
RUN cd /tmp/gromacs* && mkdir build
RUN cd /tmp/gromacs*/build && \
cmake .. -DGMX_MPI=on -DGMX_THREAD_MPI=ON -DGMX_GPU=ON -DCUDA_TOOLKIT_ROOT_DIR=/usr/local/cuda -DGMX_BUILD_OWN_FFTW=ON -DCMAKE_INSTALL_PREFIX=/opt/gromacs && \
make -j $(nproc) && make install
RUN echo "source /opt/gromacs/bin/GMXRC" >> /etc/profile
###################################################
## supervisor container startup
ADD conf/supervisord/supervisord.conf /etc/supervisor/supervisord.conf
ADD supervised-scripts/mpi-run.sh supervised-scripts/mpi-run.sh
RUN chmod 755 supervised-scripts/mpi-run.sh
EXPOSE 22
RUN export PATH="$PATH:/opt/openmpi/bin"
ADD batch-runtime-scripts/entry-point.sh batch-runtime-scripts/entry-point.sh
RUN chmod 0755 batch-runtime-scripts/entry-point.sh
CMD /batch-runtime-scripts/entry-point.sh
After the container is built, push the image to your Amazon ECR repository and note the container image URI for later steps.
Set up GROMACS

For the input files, use the Chalcone Synthase (1CGZ) example, from RCSB.org. For this post, just run a simple simulation following the Lysozyme in Water GROMACS tutorial.
Execute the production MD run before the analysis (that is, after the system is solvated, neutralized, and equilibrated), so you can show that the longest part of the simulation can be achieved in a containizered workflow.
It is possible from the tutorial to run the entire workflow from PDB preparation to solvation, and energy minimization and analysis in AWS Batch.
Set up the compute environment
For the purpose of running the MD simulation in this test case, use two p3.2xlarge instances. Each instance provides one NVIDIA Tesla V100 GPU for which GROMACS distributes the job. You don’t have to launch specific instance types. With the p3 family, the MPI-wrapper can concomitantly modify the MPI ranks to accommodate the current GPU and node topology.
When the job is executed, instantiate two MPI processes with two OpenMP threads per MPI process. For this post, launch EC2 OnDemand, using the Amazon Linux AMIs we can take advantage of per-second billing.
Under Create Compute Environment, choose a managed compute environment and provide a name, such as gromacs-gpu-ce. Attach two roles:
- AWSBatchServiceRole—Allows AWS Batch to make EC2 calls on your behalf.
- ecsInstanceRole—Allows the underlying instance to make AWS API calls.
In the next panel, specify the following field values:
- Provisioning model: EC2
- Allowed instance types: p3 family
- Minimum vCPUs: 0
- Desired vCPUs: 0
- Maximum vCPUs: 128
For Enable user-specified Ami ID and enter the AMI that you created earlier.
Finally, for the compute environment, specify the network VPC and subnets for launching the instances, as well as a security group. We recommend specifying a placement group for tightly coupled workloads for better performance. You can also create EC2 tags for the launch instances. We used name=gromacs-gpu-processor.
Next, choose Job Queues and create a gromacs-queue queue coupled with the compute environment created earlier. Set the priority to 1 and select Enable job queue.

Set up the job definition
In the job definition setup, you create a two-node group, where each node pulls the gromacs_mpi image. Because you are using the p3.2xlarge instance providing one V100 GPU per instance, your vCPU slots = 8 for scheduling purposes.
Submit the GROMACS job
In the AWS Batch job submission portal, provide a job name and select the job definition created earlier as well as the job queue. Ensure that the vCPU value is set to 8 and the Memory (MiB) value is 24000.
Under Environmental Variables, within in each node group, ensure that the keys are set correctly as follows.
| Key | Value |
| SCRATCH_DIR | /scratch |
| JOB_DIR | /efs |
| OMP_THREADS | 2 |
| GMX_COMMAND | gmx_mpi mdrun -deffnm md_0_1 -nb gpu |
| MPI_THREADS | 2 |
| S3_INPUT | s3://<your input> |
| S3_OUTPUT | s3://<your output> |
Submit the job and wait for it to enter into the RUNNING state. After the job is in the RUNNING state, select the job ID and choose Nodes.

The containers listed each write to a separate Amazon CloudWatch log stream where you can monitor the progress.
After the job is completed the entire working directory is compressed and uploaded to S3, the trajectories (*.xtc) and input .gro files can be viewed in your favorite MD analysis package. For more information about preparing a desktop, see Deploying a 4x4K, GPU-backed Linux desktop instance on AWS.
You can view the trajectories in PyMOL as well as running any subsequent trajectory analysis.
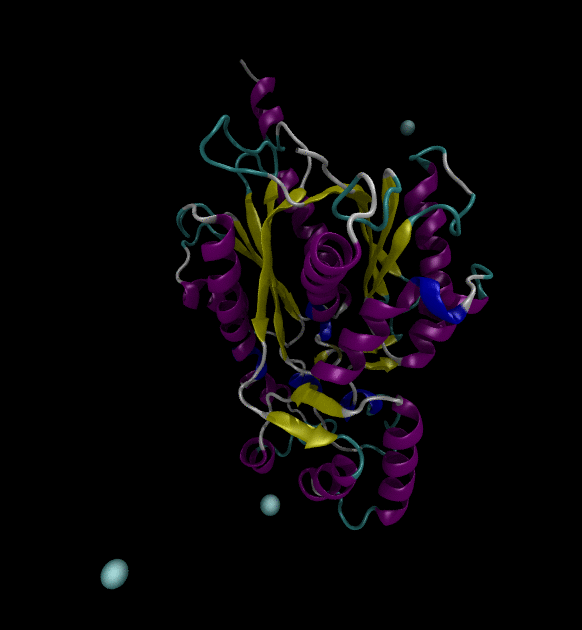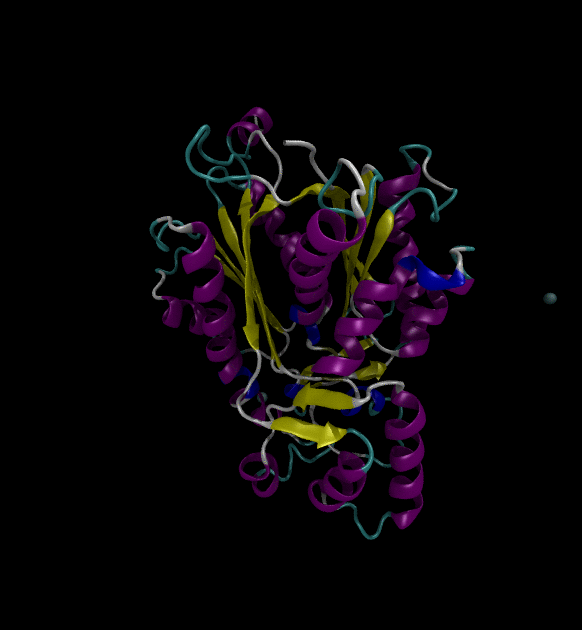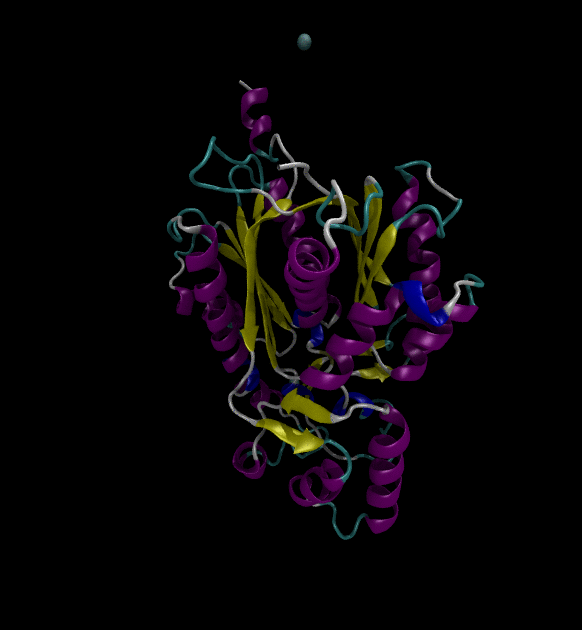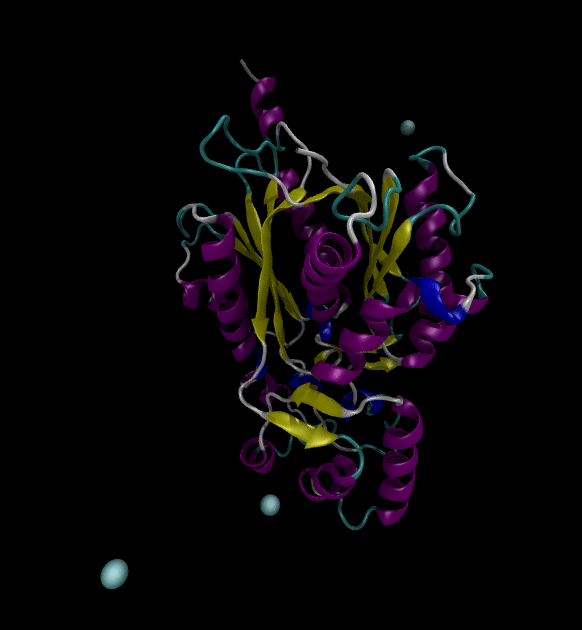
Extending the solution
As we mentioned earlier, you can take this core workload and extend it as part of a job execution chain in a workflow. Native support for job dependencies exists in AWS Batch and alternatively in AWS Step Functions. With Step Functions, you can create a decision-based workflow tree to run the preparation, solvation, energy minimization, equilibration, production MD, and analysis.

Conclusion
In this post, we showed that tightly coupled, scalable MD simulations can be executed using the recently released multi-node parallel jobs feature for AWS Batch. You finally have an end-to-end solution for distributed and MPI-based workloads.
As we mentioned earlier, many other applications can also take advantage of this feature set. We invite you to try this out and let us know how it goes.
Want to discuss how your tightly coupled workloads can benefit on AWS Batch? Contact AWS.