AWS HPC Blog
Dataset of protein-ligand complexes now available in the Registry of Open Data on AWS
 This post was contributed by U. Deva Priyakumar, Rakesh Srivatsava, Prathit Chatterjee, Vladimir Aladinskiy, Ramanathan Sethuraman, Yusong Wang, Alex Iankoulski, and Beryl Rabindran
This post was contributed by U. Deva Priyakumar, Rakesh Srivatsava, Prathit Chatterjee, Vladimir Aladinskiy, Ramanathan Sethuraman, Yusong Wang, Alex Iankoulski, and Beryl Rabindran
Today, we’re excited to announce the release of a comprehensive dataset featuring molecular dynamics (MD) trajectories for over 16,000 protein-ligand complexes (PLCs). This dataset, now available on AWS as part of the Registry of Open Data on AWS, encapsulates time-resolved data on atomic and molecular interactions within each protein-ligand system.
This represents a valuable resource for research at the intersection of machine learning and structural biology. This dataset can be applied to addressing essential challenges in modern drug design, potentially aiding the discovery of new therapeutics.
Why is this needed?
The rapid advancement of computing technologies, particularly artificial intelligence (AI), has revolutionized various domains, including drug discovery. Curated datasets are crucial for developing reliable, generalizable, and accurate models for practical applications.
However, generating experimental data on a large scale is a cost-prohibitive and arduous process. On the other hand, simulations have proven extremely valuable in life sciences and help to compensate for the lack of experimental studies. To address this need, teams from IIIT-Hyderabad, Intel, AWS, and Insilico Medicine (AWS-IIITH-Intel-Insilico – AI3), collaborated to perform massive physics-based calculations on about 20,000 protein-ligand complexes (PLCs).
This generated dataset provides dynamic, data-enriched protein-ligand coordinates and can be instrumental for training machine-learning solutions, such as artificial neural networks, to predict various aspects of ligand-protein binding.
Available now
The AI3 dataset is now accessible to the public at no cost through the AWS Open Data Sponsorship Program. AWS users will not need to pay transfer fees or long-term storage costs to access this data, or to maintain a personal copy of this data. By democratizing access to the PLC data through this collaboration, we aim to inspire innovation in computational drug discovery, thereby transform health outcomes around the world.
What’s included
The AI3 dataset contains molecular dynamics snapshots and binding energy estimates, along with individual energy components, computed for 16,692 PLCs from RCSB PDB (Research Collaboratory for Structural Bioinformatics Protein Data Bank, 2023).
The code bases come from IIIT-H (for initial input preparations), AWS (for undertaking all-atomistic simulations in GROMACS simulations package, followed by MMPBSA calculations), and Insilico Medicine (for undertaking Alchemical calculations). They have been publicly released in a GitHub repository.
As a next step, the team plans to augment the dataset by adding 10 metastable intermediates for each original PLC – making 200,000 PLC conformations in total. We will write about this again when the work is complete.
How we generated this data
Simulation of a large number of PLCs on a molecular level requires vast computational resources. Using GROMACS, a single PLC can be simulated on one compute instance using either multiple GPUs or multiple Intel Xeon CPU cores.
Depending on the number of atoms in the PLC (within a range from a few thousand to 500,000) it takes between 2 and 12 hours to complete a single run. Simulating hundreds to hundreds of thousands of PLCs goes beyond the boundaries of a single compute instance. Individual PLCs can, however, be simulated independently, making it possible to run these in parallel.
We designed the technology stack for this architecture for performance optimization and cost-efficiency, and aimed for execution times measured in hours or days. We used Intel Xeon CPUs to minimize cost and facilitate both vertical and horizontal scaling – only possible because of the large selection and availability of Amazon Elastic Compute Cloud (Amazon EC2) instance types based on Intel architectures.
For data storage, we used Amazon FSx for Lustre for short-term storage needs, and Amazon Simple Storage Service (Amazon S3) for long-term storage. We also used standard Elastic Network Interface networking in our cluster configuration.
We built our software stack on Amazon Linux 2 and encapsulated it in the Amazon Machine Image (AMI) we created for the cluster nodes. This included . To create the infrastructure, we used an open-source project called aws-do-pcluster, which allowed us to spin up AWS ParallelCluster using the do-framework.
ParallelCluster allowed us to scale to an impressive 300,000 vCPUs concurrently within a single Availability Zone, which can be further expanded by incorporating additional Availability Zones in the same region for a larger workload. This scalability enabled us to manage and process vast amounts of data efficiently, ensuring that our applications remain responsive and performant. The auto-scaling capabilities of AWS ParallelCluster allowed us to automatically adjust the number of vCPUs based on demand, ensuring cost efficiency.
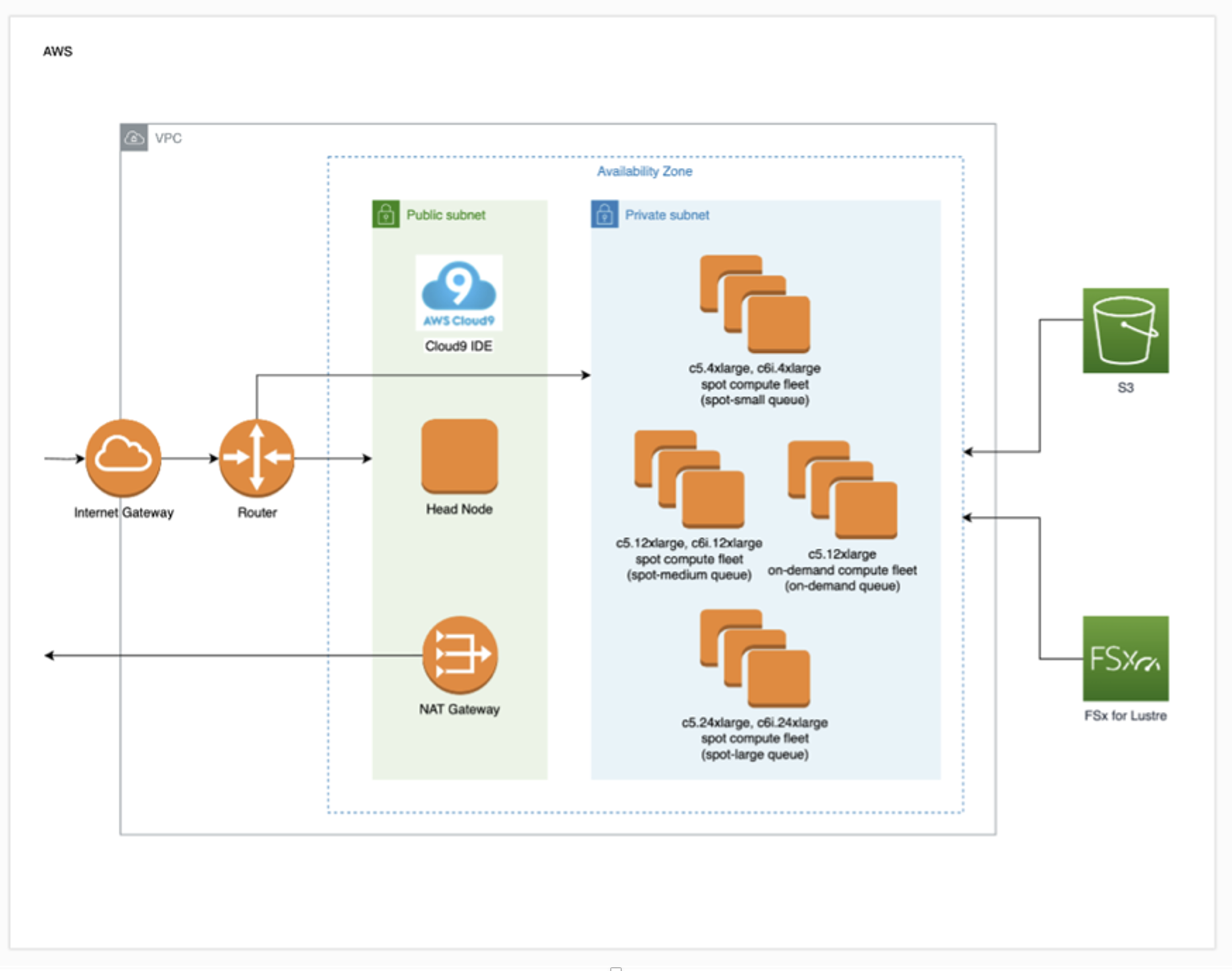
Figure 1 – PLC Deployment Architecture. The deployment architecture uses a standard Parallel Cluster within an AWS VPC. A scratch FSx for Lustre volume is mounted on the head node and all compute nodes. To minimize cost, the architecture uses primarily SPOT instances with “lowest price” allocation strategy. A partition of on-demand instances is also added to the cluster primarily for development and experimentation purposes. Each of the AWS ParallelCluster partitions shown in the private subnet corresponds to a SLURM queue. When the queue contains no jobs, the partition is automatically scaled to 0 nodes. When jobs are submitted to the SLURM queues, compute nodes of the corresponding EC2 instance types are automatically added to the cluster, thus creating an elastic compute environment.
Conclusion
Computational researchers working on drug discovery can use this data for their own workflows. That could be the analysis of changes in the shapes of proteins, comparison of binding sites, energy landscape mapping, studying the dynamics of protein-ligand interactions, or estimating binding affinity.
We’re looking forward to seeing what new and interesting questions the global computational drug discovery community will be able to answer by bringing these, and other datasets together in the cloud.
Contact us to let us know about your insights and breakthroughs using the AI3 dataset, and if the Registry of Open Data helps you along the way.
Contributors:
Insilico Medicine: Vladimir Aladinskiy, Evgeny Kirilin, Igor Diankin, Georgiy Andreev, Arkadii Lin, Eugene Babin, and Petrina Kamya
IIITH: Rakesh Srivastava, Prathit Chatterjee, Indhu Ramachandran, and U. Deva Priyakumar
Intel: Farid El Chalouhi, Chethan Rao, Arun Karthi Subramaniyan, Akanksha R Bilani, Varma S Konala, and Ramanathan Sethuraman
AWS: Alex Iankoulski, Yusong Wang, Stephen Litster, and Srinivas Tadepalli