AWS HPC Blog
Running accurate, comprehensive, and efficient genomics workflows on AWS using Illumina DRAGEN v4.0
This post was contributed by Shyamal Mehtalia, Principal Software Engineer 2 at Illumina, Duke Tran, Staff Bioinformatics Scientist at Illumina, Heejoon Jo, Senior Software Engineer at Illumina, Olivia Choudhury, PhD, Senior Partner SA at AWS, Jason McCloskey, Genomics Product SA at AWS, and Sujaya Srinivasan, Genomics SA at AWS.
Introduction
The reduced cost of DNA sequencing technology has led to an exponential growth of raw sequencing data. To keep pace with this development, secondary analysis tools that can provide fast and accurate results in a cost-effective manner are needed to extract actionable genomic insights. Illumina’s DRAGENTM (Dynamic Read Analysis for GENomics) addresses key pain points associated with next-generation sequencing (NGS) data analysis. These include a significant increase in sequencing data, limited on-premises compute options, inadequate CPU-based bioinformatics tools for rapid turnaround time, and high cost of secondary data analysis. DRAGEN empowers laboratories and organizations of all sizes to break through the bioinformatics bottleneck faster with accurate, comprehensive, and efficient secondary analysis pipelines. DRAGEN enables laboratories and researchers to do more with their genomic data by providing a robust suite of secondary analysis capabilities for a variety of experiment types, including whole genome, whole exome, RNA-Seq, methylome, and COVID detection pipelines.
DRAGEN’s DNA pipeline supports BCL to FASTQ conversion, alignment, sorting, duplicate marking, and variant calling. RNA and Methylation pipelines include RNA-Seq aligners, RNA-specific analysis, and downstream methylation calling. DRAGEN cloud offerings, powered by AWS, also include DRAGEN Apps on BaseSpace Sequence HubTM and DRAGEN pipelines on Illumina Connected AnalyticsTM.
In this blog, we provide a walkthrough of running DRAGEN on AWS and highlight our results from the DRAGEN v4.0 DNA sequencing pipeline showing that, in addition to accuracy and efficiency, it supports the most comprehensive suite of tools for DNA sequencing, including copy number analysis, structural variants, SMN callers, repeat expansion detection, and pharmacogenomics insights for complex genes. Results from these tools are available through a single call to DRAGEN with the appropriate command line options, streamlining results for customers of all disciplines. We also evaluate the performance of the suite with respect to key metrics: runtime, cost, and concordance.
Ways to Run DRAGEN
DRAGEN’s suite of secondary analysis pipelines are offered on a variety of platforms, enabling laboratories to select a solution that best fits their needs.
DRAGEN secondary analysis pipelines are available on Illumina Connected Analytics (ICA), a secure genomics data platform built to operationalize informatics and drive scientific insights. DRAGEN on ICA empowers customers to manage, analyze, and explore large volumes of data in a secure, scalable, and collaborative environment that couples the exceptional speed and accuracy of DRAGEN.
DRAGEN Bring Your Own License (BYOL) and DRAGEN on AWS Marketplace are available for laboratories looking to run the secondary analysis platform in their existing cloud environments. DRAGEN on AWS Marketplace is available for users looking to try out DRAGEN in a flexible environment with a premium pay-as-you-go model.
Multiple options for accessing DRAGEN allow labs to select the solution that best suits the type and scale of their projects. Regular and timely updates are made to DRAGEN across the various access points to provide performance improvements, enhancements, and additional functionality. This ensures that DRAGEN’s highly accurate, comprehensive, and efficient analysis pipelines are available and up-to-date for all users.
Pre-requisites
The pre-requisites for running Illumina DRAGEN on AWS include:
- Subscription to DRAGEN on Illumina Connected Analytics, BaseSpace Sequence Hub, or run directly on AWS using DRAGEN Bio-IT Platform AMI in AWS Marketplace.
If using DRAGEN AMI in AWS Marketplace, additional pre-requisites include access to an AWS account with:
- Amazon EC2 F1 instances in public regions (North Virginia, Oregon, Dublin, or Sydney)
- Permission to use Amazon S3
- [Optional] AWS QuickStart for Illumina DRAGEN
Benchmark Analysis
The DRAGEN DNA pipeline includes optimized algorithms for mapping, sorting, duplicate marking, and haplotype variant calling. It also provides lossless compression and optimized conversion of BCL files to FASTQ format. In addition to haplotype variant calling, the pipeline supports calling of copy number and structural variants, as well as detection of repeat expansions. Figure 1 presents the different processes that are available in the DRAGEN DNA pipeline.

Figure 1: DRAGEN DNA pipeline, showing FASTQ or BAM/CRAM input files being processed through a series of steps top obtain mapped reads and variant call files.
For benchmark analysis, we used the Genome in a Bottle (GIAB) samples HG002, HG003, and HG004 that were obtained from the Google Brain dataset available at the AWS Registry of Open Data (RODA) at s3://genomics-benchmark-datasets/google-brain/. The samples are 30x whole genomes aligned to the hg38 alt-masked graph v2 genome hash table from Illumina.
We ran the DNA pipeline of DRAGEN v4.0 software using FASTQ files as input to perform mapping/alignment and calling/genotyping of all variants that the pipeline is capable of, including small variant, structural variant, CYP2B6, CYP2D6, GBA, SMN, PGx Star Allele, and repeat expansion detection with an expanded catalog of Short Tandem Repeat (STR) loci available beginning with v4.0. Mapping/alignment and comprehensive variant calling were performed using a single command line that runs all the pipelines in an end-to-end workflow, thereby eliminating the need for manual step-wise processing by the user. For comparative analysis, we ran the experiment on the three samples across f1.4xlarge and f1.16xlarge EC2 instances.
Launch F1 instance with DRAGEN AMI
DRAGEN is available on AWS Marketplace as an Amazon Machine Image (AMI). An AMI provides the information and pre-requisites to launch an EC2 instance. After subscribing to the DRAGEN AMI, we launched two EC2 F1 instances: f1.4xlarge and f1.16xlarge with 500 GB root volume for storage and default VPC and subnet.
As an alternative to launching the F1 instance using the DRAGEN AMI directly via SSH, customers can also use the Illumina DRAGEN QuickStart from AWS, which deploys the necessary backend infrastructure in the customer’s account, with minimal effort from the customer. The DRAGEN QuickStart, which leverages AWS Batch, allows customers to simplify the processing of 100’s or 1000’s of samples by submitting them to a job queue. AWS Batch manages the execution of the jobs, so that the customer does not need to manage any compute clusters.
Run DRAGEN DNA Pipeline
We executed the following commands to run DRAGEN’s end-to-end alignment and variant calling pipeline. This takes the paired-end compressed FASTQ files and reference hash table as input and generates a map/aligned, sorted, and duplicate-marked CRAM file and variant calls in VCF and gVCF file formats as output. We enabled flagging of duplicate output alignment records, saving of the output from the map/align stage, small variant calling, structural variant (SV) calling, copy number variant (CNV) calling in self normalization mode, CYP2D6 diplotyping, filtering of variant calls, and repeat genotyping (or expansion detection) using the corresponding flags: enable-duplicate-marking, enable-map-align-output, enable-variant-caller, enable-sv, enable-cnv, cnv-enable-self-normalization, enable-cyp2d6, vc-hard-filter, repeat-genotype-enable, respectively. Although we tested DRAGEN with BYOL option, customers can also use DRAGEN from AWS Marketplace to run these experiments.
The following is the DRAGEN command line used for the HG002 GIAB sample:
The other GIAB samples, HG003 and HG004, were processed in a similar manner.
Results
For benchmark analysis, we ran the DRAGEN v4.0 DNA pipeline on the three GIAB datasets (HG002, HG003, and HG004), as discussed above. To compare the evaluation metrics, i.e., runtime, concordance, and cost, we conducted the experiments on two types of EC2 instances: f1.4xlarge and f1.16xlarge. The f1.4xlarge instance consists of 2 FPGAs, 8 cores, 16 vCPUs, 244 GB instance memory, and 940 GB SSD storage, whereas the f1.16xlarge instance consists of 8 FPGAs, 32 cores, 64 vCPUs, 976 GB instance memory, and 4 x 940 GB SSD storage.
Figure 2 presents the total runtime incurred by running the DRAGEN v4.0 DNA pipeline for the given dataset and EC2 instance types in the us-east-1 region. The runtime is further broken down into individual steps involved in the pipeline. As seen in the figure, the total runtime for the pipeline on f1.4xlarge is ~95 mins, and ~48 mins on f1.16xlarge. This is significantly faster than the GATK best practices pipeline [1], and includes a more comprehensive set of analysis, including all callers, copy number variants, structural variants, repeat genotyping, targeted callers CYP2B6, CYP2D6, GBA, SMN, and PGx Star Allele callers. Total runtime also included generation of quality control metrics using a hg38 graph-based reference genome. As of December 2022, the compute cost incurred in running this workload by the f1.4xlarge and f1.16xlarge on-demand instances in us-east-1 region are $6.60 and $13.20, respectively. This indicates that a majority of such workloads can be run on f1.4xlarge instances in a cost-efficient manner.

Figure 2: Runtime (mins) incurred by DRAGEN v4.0 DNA pipeline tested on GIAB samples across f1.4xlarge and f1.16xlarge EC2 instances. Note that runtimes for individual steps may be concurrent and are not necessarily additive to the total runtime as a whole.
We also looked at the concordance of the small variants (SNPs and Indels) against the truth set from the GIAB Consortium [2]. We used the best practices outlined in [3] to compute precision, recall, and F1-score across the results obtained from all 3 samples on both EC2 instance types (Figure 3). In order to compute concordance, we used truth set VCFs from here. The hap.py tool was used with the RTG tools VCFeval engine to compute concordance as follows:
Figure 3 showcases DRAGEN’s award-winning high accuracy across all three samples tested. Furthermore, the concordance metrics are consistent across both f1.4xlarge and f1.16xlarge EC2 instances. These results show that DRAGEN’s performance is consistent and deterministic across runs, samples, and FPGA instance types.
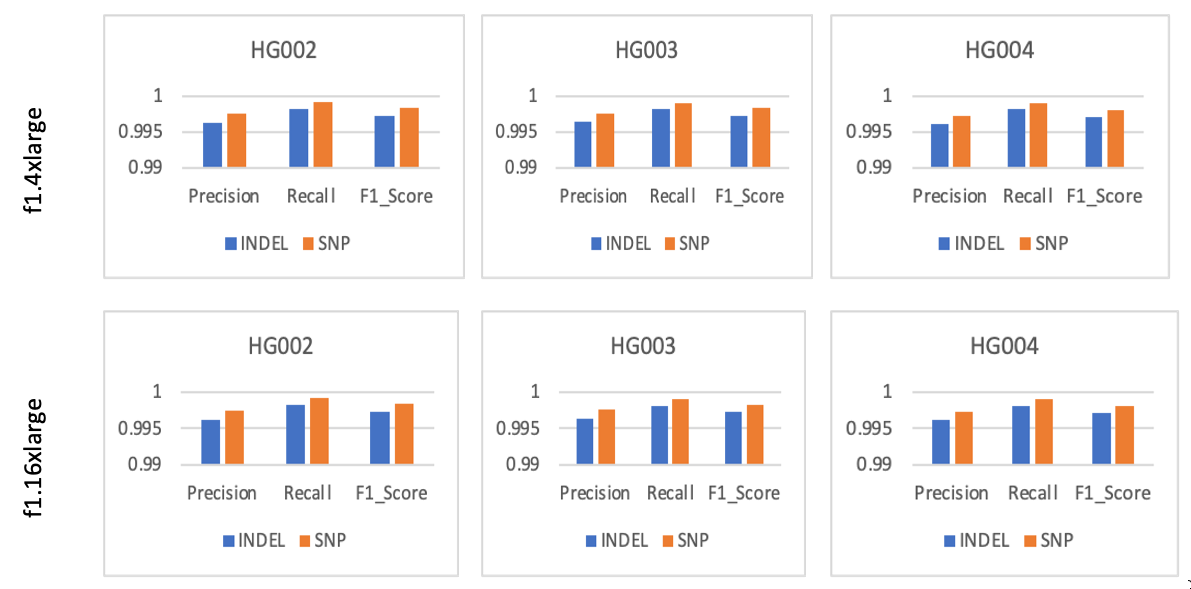
Figure 3: Precision, Recall, and F1 score for variants (indel, SNP) tested on GIAB samples across f1.4xlarge and f1.16xlarge EC2 instances. The results show high (greater than 99%) precision, recall, and model accuracy for both insertion-deletion and SNPs.
Cleaning Up
If running DRAGEN in your own environment, terminate all the resources created in the steps above to avoid incurring future charges.
Conclusion
The DRAGEN v4.0 DNA pipeline was run for three samples (GIAB HG002, HG003, and HG004) across two EC2 instance types (fx1.4xlarge, f1.16xlarge) after which cost, efficiency, and concordance of small variants were measured. DRAGEN v4.0 demonstrated exceptional accuracy and efficiency across samples and FPGA instance types. The DRAGEN v4.0 DNA pipeline completed analysis and QC metrics for all callers and an extensive number of additional targeted callers within ~95 mins or ~48 mins (f1.4xlarge or f1.16xlarge, respectively), representing a significantly faster and more comprehensive analysis compared to the GATK best practices pipeline [1]. High concordance was also demonstrated, with DRAGEN v4.0 consistently showing the same award-winning accuracy across runs, samples, and instances.
The content and opinions in this blog are those of the third-party author and AWS is not responsible for the content or accuracy of this blog.
Customer Success Stories
Find out more about how customers are using DRAGEN to accelerate genomic insights –
- Munich Leukemia Laboratory – Munich Leukemia Laboratory uses DRAGEN on AWS Infrastructure to reduce turnaround time from 24 hours to 4 hours.
- UMCCR Genomics Platform Group – DRAGEN pipelines on Illumina Connected Analytics enable the Genomics Platform Group to digitally transform their sequencing workflows.
- Basepaws – Basepaws, a Zoetis company, leverages the Illumina DRAGEN Bio-IT Platform to develop direct-to-consumer DNA kits that provide cat owners with insights about their pets’ health.
References:
- Betschart, Raphael, et al. “Comparison of calling pipelines for whole genome sequencing: an empirical study demonstrating the importance of mapping and alignment.” bioRxiv (2022).
- Zook, J.M., McDaniel, J., Olson, N.D. et al. An open resource for accurately benchmarking small variant and reference calls. Nat Biotechnol 37, 561–566 (2019). https://doi.org/10.1038/s41587-019-0074-6
- Krusche, P., Trigg, L., Boutros, P.C. et al. Best practices for benchmarking germline small-variant calls in human genomes. Nat Biotechnol 37, 555–560 (2019). https://doi.org/10.1038/s41587-019-0054-x