AWS for Industries
Hummingbird – a tool for effective prediction of performance and costs of genomics workloads on AWS
Blog guest authored by Utsab Ray, Amir Alavi, Amit Dixit, Vandhana Krishnan, and Amir Bahmani from Stanford University.
Genomics researchers often face challenges in accurately estimating the compute and memory resources required for their workloads as they work to migrate their data processing to the cloud. AWS cloud computing infrastructure offers scalable and cost-effective solutions for genomics workloads with more than 270 instance options to choose from. Since cloud costs are directly tied to the size of compute resources and the time duration of usage, overprovisioning of resources can lead to higher costs, thereby preventing researchers from using the cloud effectively.
Hummingbird is a tool created at Stanford University that helps researchers effectively profile and predict the performance of a genomics tool or workflow. It provides recommendations on the fastest, cheapest and most cost-effective Amazon EC2 instance types for their particular use case. Hummingbird [1] is an open-source tool that is freely available at https://github.com/StanfordBioinformatics/Hummingbird.
Researchers need insights into cost and runtime forecasting, cost-time trade-offs, and appropriate AWS offerings for specific jobs. While AWS offers many tools for predicting and managing costs, such as cost calculators, AWS Trusted Advisor, Cost Explorer, etc., these tools are not exclusive to advanced research areas like genomics. Hummingbird helps address these researcher needs. Additionally, when using services like AWS Batch that provision EC2 instances for jobs, researchers can use Hummingbird to determine the compute and memory requirements optimized for their workloads so that resources are right-sized and costs are kept under control.
In the words of Michael Snyder, Chair and Professor of Genetics at Stanford University, “To deliver affordable precision medicine, researchers and physicians require fast and cost-efficient systems that are able to scale efficiently. Hummingbird helps with the selection of resource-matching compute instance types on cloud platforms and can significantly reduce computational costs.”
Philip Tsao, Associate Chief of Staff for Precision Medicine, VA Palo Alto Health Care System, and Stanford Professor of Medicine, says, “VA MVP is one of the world’s largest genomic programs established in a healthcare setting. Since launching in 2011, over 850,000 Veteran partners have joined the program. As a cloud customer, we quickly realized that in order to run genomics pipelines efficiently at scale, a better understanding of each stage of a genomics pipeline is essential; therefore, in collaboration with Stanford University, we developed Hummingbird.”
In this blog post, we discuss our results from profiling popular genomics tools, like BWA-MEM and GATK HaplotypeCaller, on AWS using Hummingbird. We also demonstrate how to use Hummingbird to estimate optimal resources for other genomics workflows.
Profiling with Hummingbird in your AWS account
You can use Hummingbird for profiling genomics workloads with an AWS account, preferably with power user access, which is required in order to fully leverage the capabilities of the tool. You can deploy Hummingbird into your AWS account and begin running profiling tasks from an EC2 instance (i.e., the “Hummingbird Management Node”) by deploying an AWS CloudFormation stack using the template referenced in the following section. This stack also includes networking components (a VPC, public and private subnets, a security group, gateways, etc.) used to secure access to the management node, as well as a Network Load Balancer and Auto Scaling group that ensure that the management node is highly available.
Figure 1 illustrates the main components that are deployed by the template:
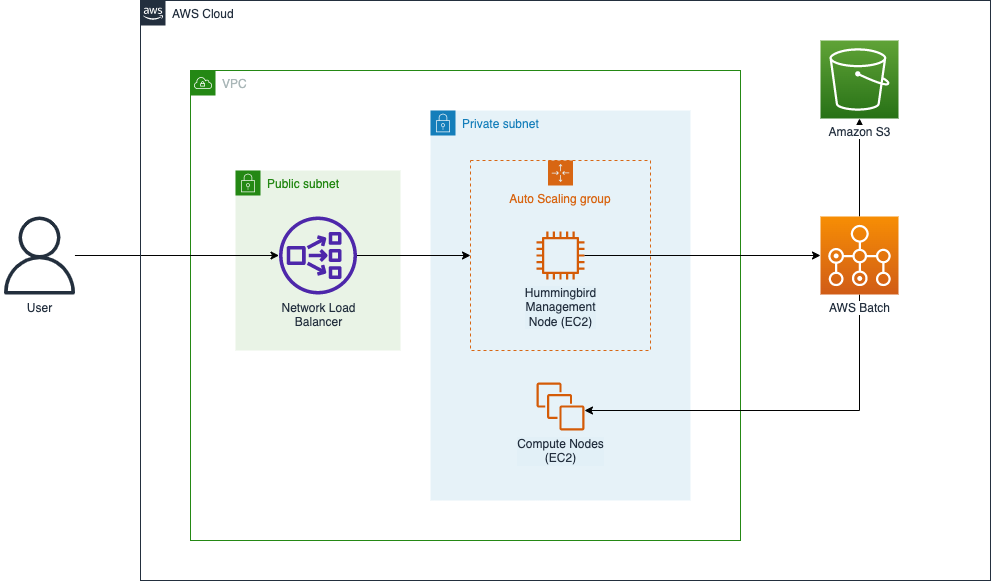
Figure 1: The Hummingbird CloudFormation stack on AWS
Once the CloudFormation stack has been deployed, you can access the Hummingbird management node using an SSH client from your local workstation. The management node will have the latest version of Hummingbird installed, along with its required software packages.
When you run Hummingbird from the management node, Hummingbird uses AWS Batch to deploy and manage EC2 instances for running the profiling tasks. Data required for the tasks is loaded from Amazon S3, and profiling results are written into S3 as profiling tasks complete. Since Hummingbird uses S3 for both task input and writing profiling results, the provided CloudFormation template includes broad, permissive access to S3 from EC2 instances launched by AWS Batch (see the ECSInstanceRole and ECSTaskExecutionRole resources in the template). Therefore, if the AWS account that you’re running Hummingbird in is used for other AWS workloads, you should ensure that you’re comfortable with Hummingbird’s broad access to S3 or modify the aforementioned IAM roles used by Hummingbird to ensure that its access is limited to the input and output buckets that you specify when you configure Hummingbird.
Steps to set up and run Hummingbird
1) Using your browser, sign in to the AWS account that you wish to deploy Hummingbird into.
2) Deploy the CloudFormation template containing the above stack by selecting the Launch Stack button.

3) Choose a key pair that you will use to SSH into the EC2 instance. If you don’t have a key pair to use, you can create one using the EC2 console.
4) Once the stack has deployed, note the domain name of the Network Load Balancer (NLB), as you will use this domain name for logging in to the Hummingbird management node.
5) SSH into the instance running Hummingbird by executing:
ssh -i <key pair file> ec2-user@<NLB domain name>
6) Go to the Hummingbird directory:
cd /usr/local/lib/python3.7/site-packages/Hummingbird
7) Edit the configuration files from the conf/examples directory and copy them to the Hummingbird directory. For example, to run BWA on AWS, edit the bwa.aws.conf.json file as shown below. A detailed explanation of the various fields is shown in the table below.
8) Execute Hummingbird:
hummingbird <conf file>
Example contents of bwa.aws.conf.json file:
A description of the fields that are supported for configuring Hummingbird:
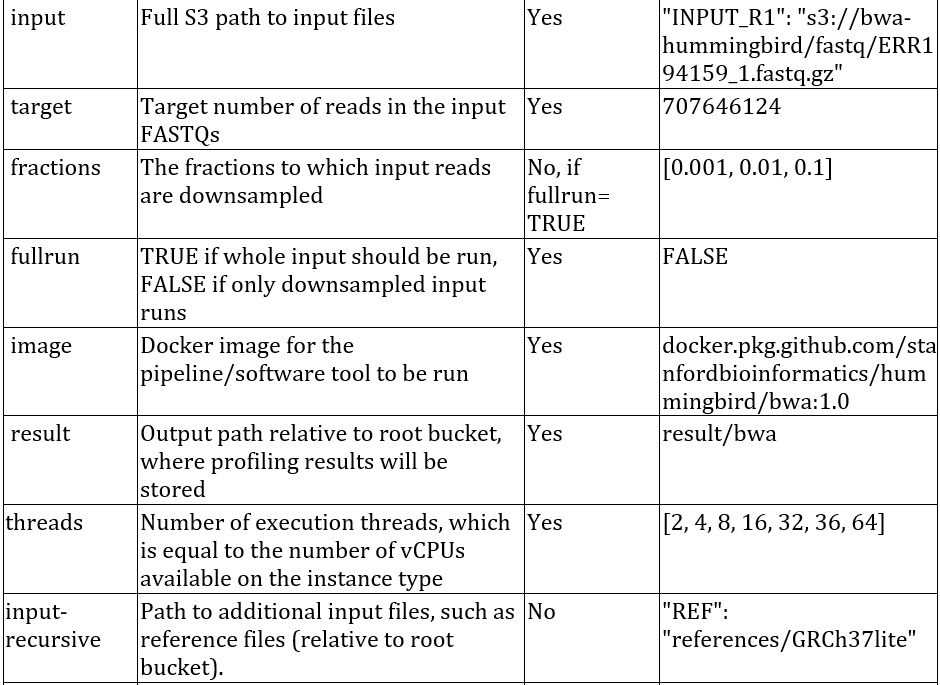

Data and tools
We set out to demonstrate Hummingbird using two popular genomics tools:
1) BWA-MEM [3] used to align DNA sequencing reads to the human reference genome.
2) HaplotypeCaller [4], which is part of the Genome Analysis Toolkit (GATK) from the Broad Institute, used to call germline single nucleotide variants (SNVs) and short insertions and deletions (INDELs).
We used FASTQ files for HG001 (NovaSeq, PCR-Free, 30x coverage) [2] from the publicly available S3 bucket (s3://genomics-benchmark-datasets/google-brain/fastq/novaseq/wgs_pcr_free) and the human reference genome files at s3://broad-references.
Results
In order to understand the impact of data availability on performance, we ran Hummingbird using the full input data as well as randomly downsampled data corresponding to 0.1%, 1%, and 10% of the input to profile the performance of BWA-MEM and HaplotypeCaller in 7 AWS instance families (c5, c5a, c5ad, c5d, i3, m5, r5).
Figure 2 shows the execution times and costs of running BWA on different instance types. The x- and y-axes denote the cost in dollars, and execution time in seconds. It is clear that the c5 family performs well in contrast to the i3 instance family. This indicates BWA-MEM is a compute and memory intensive tool but not particularly I/O intensive, so using an i3 instance type for this workload is not optimal. On the other hand, the c5, c5a, c5ad, c5d instance families have a ratio of 1:2 for vCPUs to memory, which seems to be optimal for BWA.
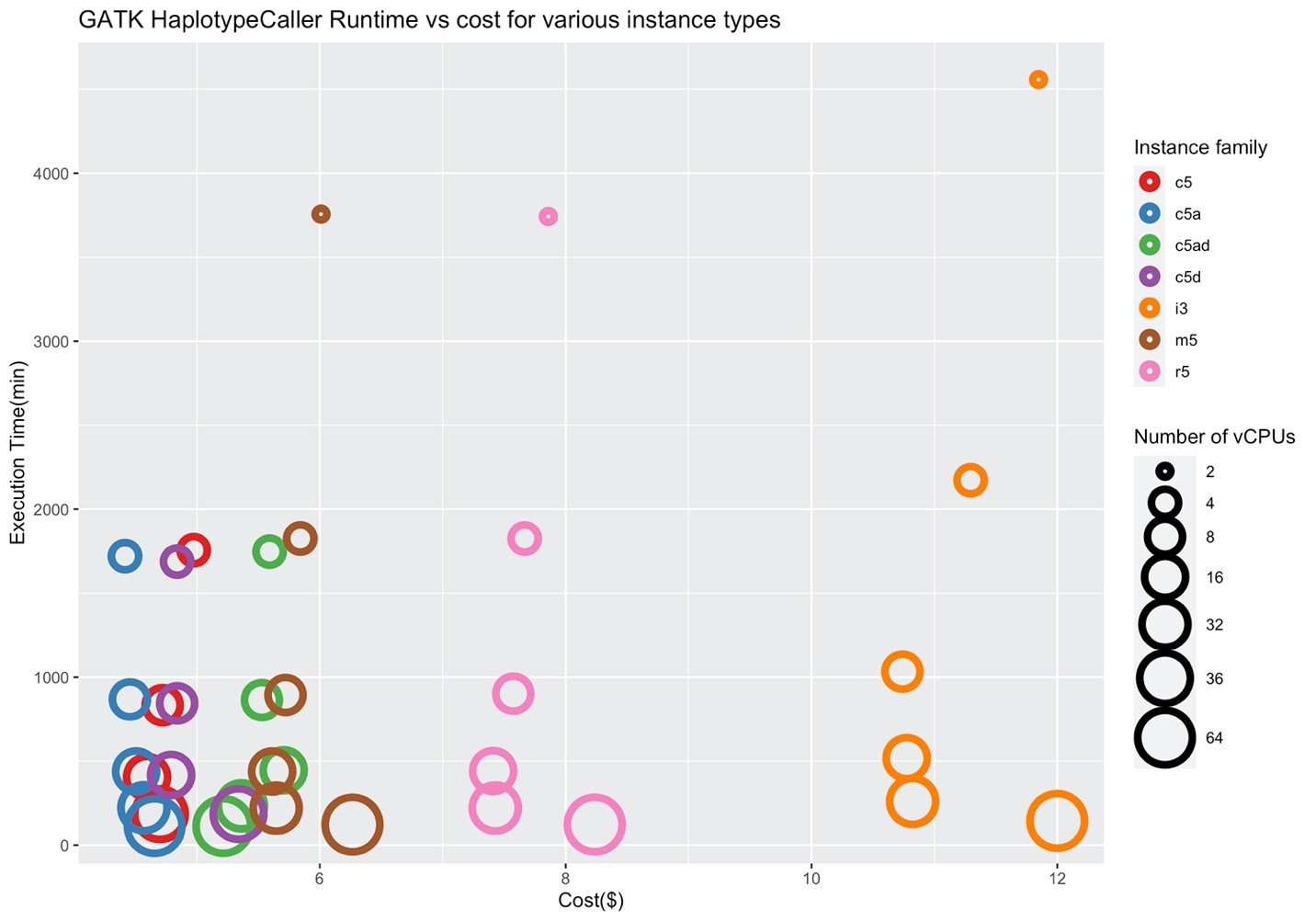 Figure 2: Price/performance across different AWS families for BWA-MEM
Figure 2: Price/performance across different AWS families for BWA-MEM
Often researchers are interested in understanding if increasing instance size helps with cost savings through decreased execution time. So, we defined a new metric, “speedup,” for measuring the relative performance of two instances processing the same workload. For example, if running BWA-MEM for a given input takes 240 seconds on c5.xlarge instance with 2 vCPUs, and the same takes 180 seconds on c5.2xlarge instance with 4 vCPUs, then speedup is S=240s/180s= 1.5. Ideal speedup implies that doubling the number of resources doubles the program speed. For this example, because we doubled the number of vCPUs, the ideal speedup is 2.
Figure 3 shows the ideal speedup, the speedup for running BWA on a downsampled set of inputs (0.1%, 1%, 10%), and the whole input file. The x- and y-axes denote the instance type and speedup. By comparing the whole input speedup and ideal speedup, we observe that BWA scales well on increasing instance sizes (i.e., increasing the number of vCPUs and the corresponding main memory). From this experiment, we can also infer that 10% downsampled input closely resembles the execution behavior of the whole input.
While we do not report the results in this blog, BWA-MEM was also run on different sequence coverages (40X, 50X), but the performance did not vary across these datasets.
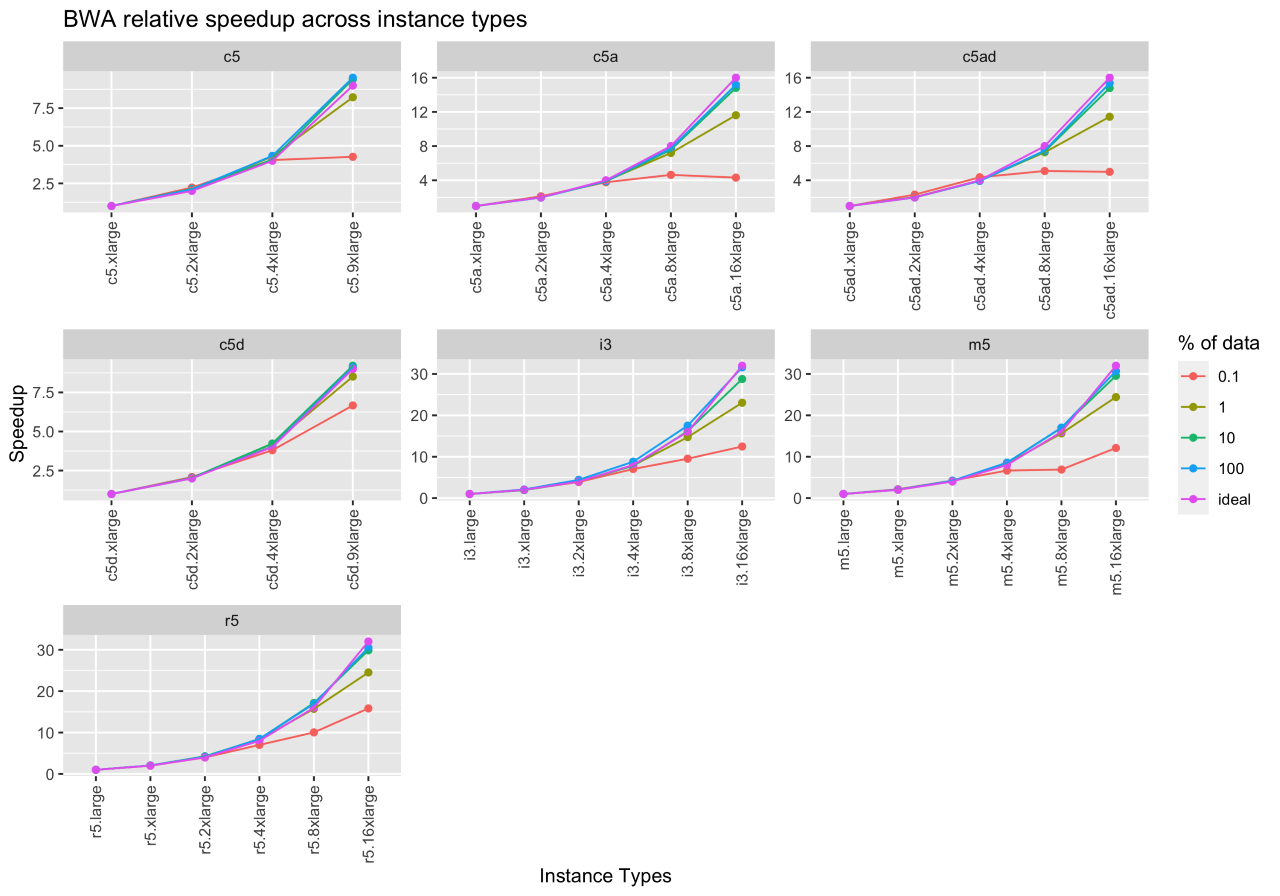 Figure 3: BWA-MEM relative and ideal speedup across EC2 instance types
Figure 3: BWA-MEM relative and ideal speedup across EC2 instance types
Figures 4 and 5 show the corresponding price/performance and relative speedup for GATK HaplotypeCaller. We can observe that the cost increases by increasing the size of the instance. Similar to BWA, I/O performance is not a key player for the performance of GATK HaplotypeCaller, and the c5 family performs better in contrast to the i3 family. Note that the c5.large, c5d.large, c5a.large, c5ad.large instances (i.e., ones with two vCPUs) were pruned by Hummingbird due to insufficient memory.
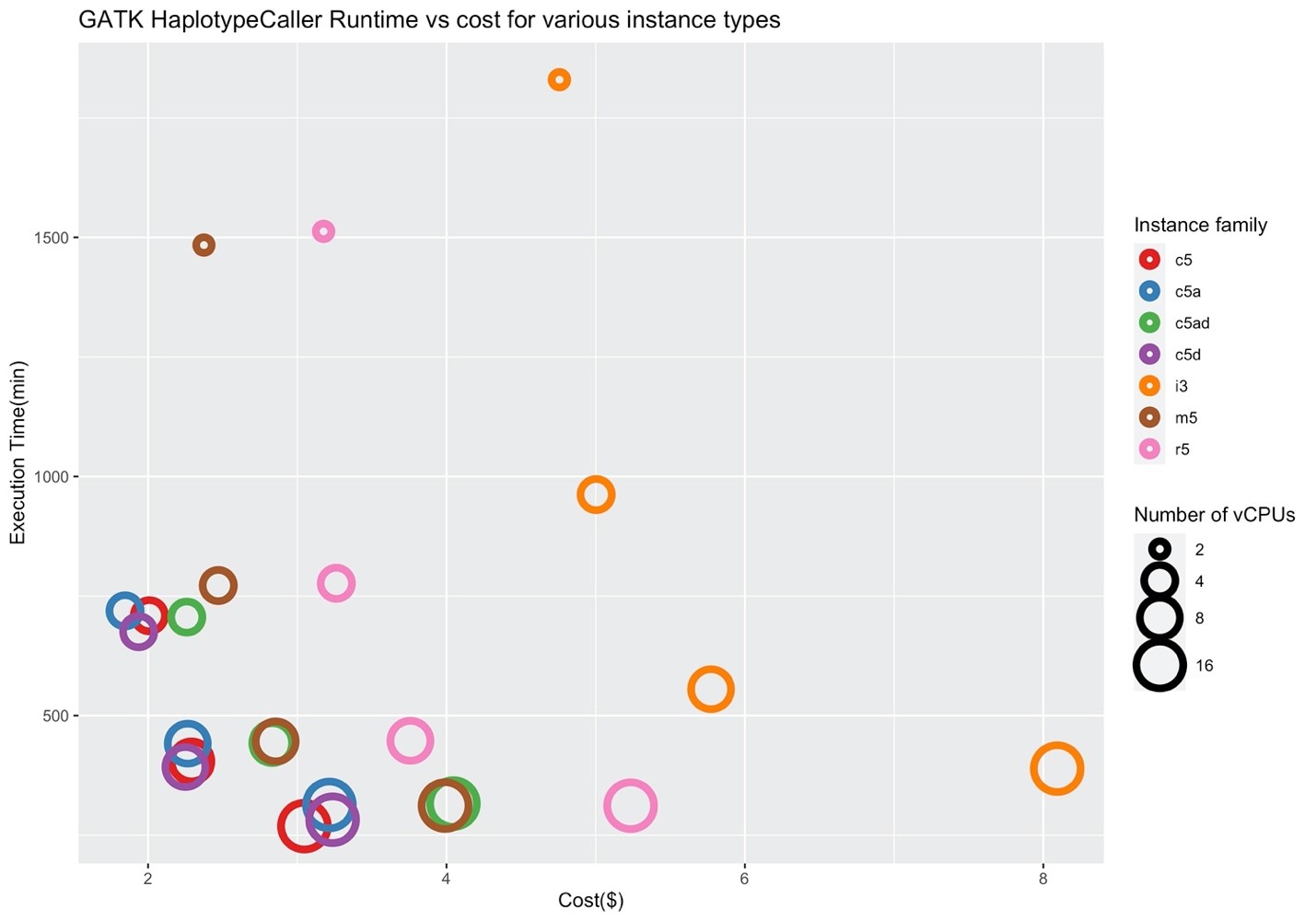
Figure 4: Price/performance across different AWS families for GATK HaplotypeCaller
Figure 5 depicts the ideal speedup and the speedup for running GATK HaplotypeCaller on a downsampled set of inputs (0.1%, 1%, 10%) and the whole input file. We observe that, unlike BWA-MEM, GATK HaplotypeCaller does not scale well on increasing instance sizes. We can observe this in Figure 4, where larger instances of the family are more expensive compared to the smaller instances due to lower efficiency. Similar to the BWA-MEM experiment, .xlarge instances were pruned due to insufficient memory.
 Figure 5: Relative and ideal speedup across instance types for GATK HaplotypeCaller
Figure 5: Relative and ideal speedup across instance types for GATK HaplotypeCaller
Conclusion
In this blog post, we demonstrated that Hummingbird works with a variety of Amazon EC2 instance families on two popular genomics tools, BWA-MEM and GATK HaplotypeCaller. By running Hummingbird on genomics tools of choice, researchers can get recommendations on the optimal instance types for performance, cost, and resource utilization. Additionally, researchers can use the recommendations from Hummingbird to determine the resource requirements (CPU and memory) for their tools and identify the instance families and the ratio of CPU to memory that work well for their needs. They could use this recommendation to define the instance types for their compute environment in AWS Batch (or context in the Amazon Genomics CLI).
Try Hummingbird for your own pipelines and report your experiences and results in the GitHub repository at https://github.com/StanfordBioinformatics/Hummingbird.
References:
[1] Bahmani, A., Xing, Z., Krishnan, V., Ray, U., Mueller, F., Alavi, A., … & Pan, C. (2021). Hummingbird: efficient performance prediction for executing genomic applications in the cloud. Bioinformatics, 37(17), 2537-2543. https://doi.org/10.1093/bioinformatics/btab161
[2] Baid, G., Nattestad, M., Kolesnikov, A., Goel, S., Yang, H., Chang, P. C., & Carroll, A. (2020). An extensive sequence dataset of gold-standard samples for benchmarking and development. bioRxiv.
[3] Li H. (2013) Aligning sequence reads, clone sequences, and assembly contigs with bwa-mem. arXiv preprint arXiv:1303.3997.
[4] McKenna A. et al. (2010) The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303.

Utsab Ray is a Software Developer at the Stanford Healthcare Innovation Lab where he focuses on the execution and optimization of genomics applications in the cloud. Prior to joining Stanford he completed his Master’s in Computer Science from NC State University.

Amir Alavi is a Software Engineer with over a decade of experience delivering large-scale software projects for enterprises and startups. Amir has a keen interest in developing design patterns and best-practices for cloud native applications.

Amit Dixit is an AWS solutions architecture consultant at the Stanford Healthcare Innovation Lab where he focuses on the development and security of genomics applications in the cloud.

Vandhana Krishnan is a senior computational and bioinformatics scientist at the Stanford Center for Genomics and Personalized Medicine. She works with the R&D teams in the Stanford Healthcare Innovation Lab, Bioinformatics-as-a-Service, Stanford Genomics, Veterans Affairs Palo Alto Health Care System, and the Clinical Genomics Program at Stanford Health Care. She builds reproducible, cost-effective, and scalable solutions employing cutting-edge technologies that aid in discoveries for various multi-omics projects.

Amir Bahmani is the Director of Stanford Deep Data Research Computing Center (DDRCC), the Director of Science and Technology at Stanford Healthcare Innovation Lab (SHIL) and a lecturer at Stanford University. He has been working on distributed and parallel computing applications since 2008. Currently, Amir is an active researcher in the VA Million Veteran Program (MVP), Human Tumor Atlas Network (HTAN), the Human BioMolecular Atlas Program (HuBMAP), Stanford Metabolic Health Center (MHC) and Integrated Personal Omics Profiling (iPOP).