AWS for Industries
DRAGEN reanalysis of the 1000 Genomes Dataset now available on the Registry of Open Data
Guest authored by Bryan Lajoie, Staff Bioinformatics Scientist at Illumina Inc.
—
We are pleased to announce the release of a comprehensive reanalysis of 3,202 deeply-sequenced samples from the 1000 Genomes Project(1kGP) using the Illumina DRAGEN (Dynamic Read Analysis for GENomics) Bio-IT platform. This seminal dataset will be freely available for researchers across the world to use as part of their genomics analysis. The alignment files and small variant, copy-number, structural, and short-tandem repeat variant calls are now available at no cost through the Registry of Open Data on AWS.
This release (1kGP-DRAGEN) includes 2,504 unrelated samples from the 1000 Genomes Project phase 3 as well as an additional 698 related samples that complete 535 mother-father-child triads, funded by the NHGRI. The samples were all sequenced at >30x coverage using the Illumina NovaSeq 6000 system with 2x150bp reads. All 3,202 samples were re-realigned to hg38 using Illumina DRAGEN v3.5.7b, powered by the Illumina Analytics Platform (IAP) and AWS.
In this post, we provide an overview of the dataset, the methods to generate the data, and how you can quickly get started.
The 1000 Genomes Project
An international effort that began in 2008, 1kGP addressed a community need to identify and catalog normal genetic variation across diverse populations. In its first phase, the 1kGP consisted of 1092 individuals from 14 subpopulations in Africa, Asia, Europe, and the Americas. A second phase of sequencing increased the dataset to 2504 individuals from 26 populations. Most recently, the genomes for another 698 related individuals were released, completing 535 parent-child triads, as well as other non-triad pedigrees.
The 1kGP remains one of the most diverse and widely used references for naturally occurring genetic variation in human populations. With over 5400 citations in the scientific literature to date, the 1kGP is regarded as “an essential tool” for genomic studies ranging from the evolutionary history of humankind to elucidating molecular mechanisms such as linkage disequilibrium and genetic recombination to the discovery and validation of disease-associated genetic mutations. For example, 1kGP data played an important role in the development of a sequence-based tool that identifies copy number variation in the SMN1 and SMN2 genes, which are used as genetic markers for Spinal Muscular Atrophy (SMA) risk. Researchers used 1kGP data to assess SMN1 and SMN2 variants copy number variation, and the identification of variant sites that can be used to reliably differentiate the two close paralogs SMN1 and SMN2 across all populations (Figure 1).

Figure 1: Distribution of SMN1, SMN2 and SMN2∆7-8 (a truncated form of SMN2) copy numbers across five populations in 1kGP. Having zero copies of SMN1 indicates a high risk for SMA. One of SMN1 indicates an SMA carrier. The copy number of SMN2 is important for clinical classification and prognosis for SMA patients.
All raw sequence data for these 1kGP samples are kept current and publicly accessible on the Registry of Open Data on AWS.
DRAGEN and AWS – Collaborating since 2016
The Illumina DRAGEN (Dynamic Read Analysis for GENomics) Bio-IT platform provides secondary analysis of NGS data from genomes, exomes, methylome, and transcriptomes. The DRAGEN Platform is a combined hardware and software solution comprised of field-programmable gate array (FPGA) accelerated algorithms. Ultra-rapid run times and uncompromising accuracy help reduce the bottleneck of secondary data analysis.
DRAGEN has been running on AWS since 2016, when Edico Genome (acquired by Illumina in 2018) released it alongside the release of the Amazon EC2 F1 instance family. In 2017, DRAGEN set a Guiness World Record for the fastest analysis of 1000 whole human genomes, completed using 1000 f1.2xlarge EC2 instances in a single AWS Region at an average compute cost under $3 genome. Since then, DRAGEN has continued to see improvements in speed, features, and quality of variant calls.
DRAGEN has continually scored highly in the PrecisionFDA Truth Challenges, and placed first in recent challenges related in regions that are difficult to map to. DRAGEN had 28% and 38% fewer errors made than the closest competitor in all-benchmark regions and difficult-to-map regions, respectively, for Illumina data (Figure 2, See the results of the challenge and learn more here).

Figure 2: Bar plot demonstrating DRAGEN accuracy (depicted by the number of false-negative (FN) and false-positive (FP), less is better) as compared to the other leading entries in the PrecisionFDA Truth Challenge V2. The DRAGEN team took 1st place in the challenge in two categories: calling variants on Illumina sequencing data in Difficult-to-Map Regions (shown here) and All Benchmark Regions (data not shown). DRAGEN++ (left most bar plot) performed best by a comfortable margin, with 28% fewer missed calls in all benchmark regions and 38% fewer in difficult-to-map areas. Plot from https://precision.fda.gov/challenges/10/view/results.
DRAGEN is capable of analyzing a ~30x human whole genome by mapping and aligning of reads, and calling single nucleotide variants (SNV), small insertion and deletions (INDELS), copy number variants (CNV), structural variants (SV), short tandem repeats (STR) and runs of homozygosity (ROH) in <120 minutes using a standard AWS F1.4x instance with only 8 CPU cores (FIGURE 3, green bar). The recently released DRAGEN v3.6.3 brings a significant improvement to run-times, enabled by innovations such as a De Bruijn graph offload to FPGA, lowering the total runtime to <80 minutes (Figure 3, red bar). For those customers unwilling to compromise on speed, DRAGEN v3.6.3, run on an AWS F1.16x lowers the runtime to <40 minutes (FIGURE 3, orange bar), providing out of the box capabilities that are complementary to rapid-sample-to-answer type use-cases. DRAGEN on AWS FPGA minimizes the need and burden of orchestrating analysis across various nodes to achieve comparable run-times.
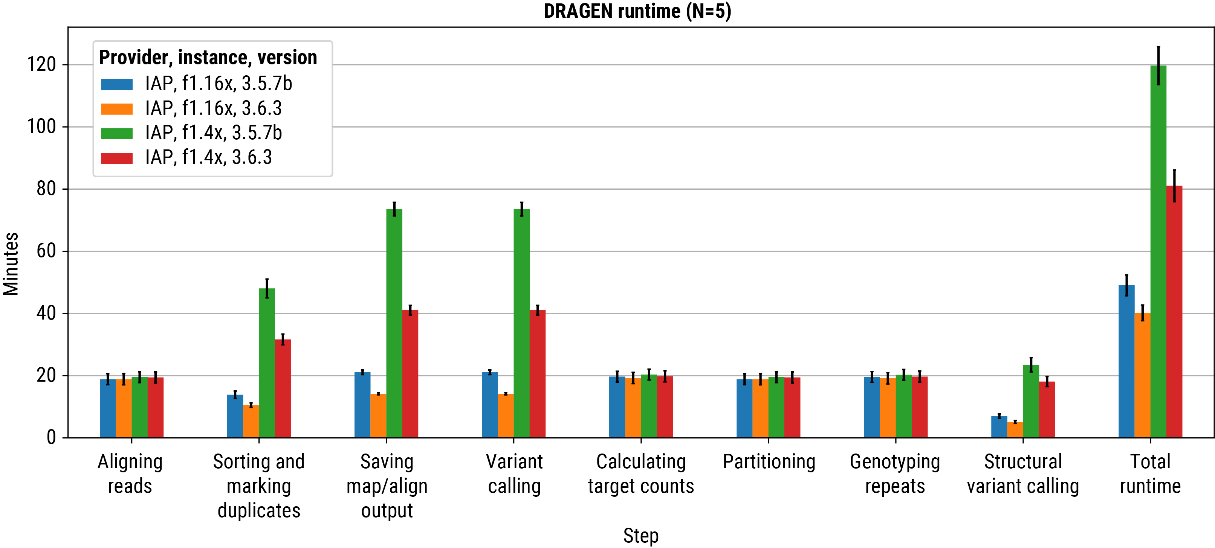
Figure 3: Bar plot demonstrating DRAGEN run-time from a representative set of ~30X 1000genomes 2,504 high-depth samples. The y-axis shows the time, and the x-axis stratifies by the ‘step’ within the DRAGEN germline pipeline. Each color bar represents a different combination of the provider (cloud platform), size (resource-type), and version (DRAGEN software). Here, “IAP” represents the Illumina Analytics Platform running on AWS. (Note, step timings are not additive as many steps run in parallel on the f1.4x node). The final ‘step’ on the far-right (total runtime) captures the all-in-one E2E performance of DRAGEN to map-align and variant-call on a set of WGS samples. Variant-calling include all major types (SNV, INDEL, CNV, SV, STR, and ROH).
Additionally, DRAGEN can easily scale using AWS Batch, which enables parallel processing of hundreds of samples. In close cooperation with AWS, DRAGEN is available to use as a Private and Public AMI, in AWS Marketplace, on Illumina’s BaseSpace Sequence Hub and soon on Illumina Analytics Platform (IAP – more information below). The AWS and DRAGEN teams have collaborated on AWS Quick Start, which helps many AWS users easily deploy and scale in their environments.
The 1KGP-DRAGEN Release
This dataset release consists of a total of 3,202 WGS, each processed independently by DRAGEN v3.5.7b, on the Illumina Analytics Platform (IAP) using Amazon EC2 f1.4xlarge instances. IAP is a scalable, secure, and compliant system which delivers DRAGEN pipelines, advanced data management, robust data science tools, and workflow automation capabilities in the cloud. IAP will be generally available in early 2021.
To highlight the diversity of the 1kg samples combined with the accuracy and completeness of DRAGEN germline, we explored some of the DRAGEN metrics produced across the 3,202 samples (Figure 4). All samples were sequenced to a >30x depth (35.6x +- 3.86x), and run-times scaled linearly with the depth (Figure 4 top left and top right panel). To highlight the completeness of the DRAGEN solution, the total number of SNV/INDEL, CNVs, and SVs were plotted for each sample, stratified by population and colored by super-population (Figure 4, bottom row).

Figure 4: A subset of DRAGEN metrics plotted against 1KG populations. The 1KG samples are colored by the 5 major super-populations (AFR, EUR, SAS, EAS, AMR). The x-axis is ordered by the super-population and stratified by the population. Top row, from left to right: Average Coverage (35.6x +/- 3.86x). Het/Hom captures notable ancestry dependent features and is used as a QC check. Total Runtime (normalized to 30x depth) demonstrates the speed at which DRAGEN can map-align, and variant call across ALL variant classes (all populations, 114.98m +/- 5.05m). AFR samples in blue demonstrate a slightly higher runtime, due to the hg38 reference bias (120.07m +/- 3.96). Bottom Row, from left to right: Number of Small Variants (single nucleotide variant (SNV) and INDEL). Number of Copy Number Variants (CNV, amplifications & deletion). Number of Structural Variants (SV, deletions, insertions and duplications).
To facilitate a more streamlined workflow, cram files from 1kg were first converted into BAM format, and then map-aligned and variant-called on DRAGEN 3.5.7b using the below cmd/options.
All data was aligned to the hg38_alt_aware_nohla v8 DRAGEN hash-table (https://1000genomes-dragen.s3.amazonaws.com/reference/hg38_altaware_nohla-cnv-anchored.v8.tar) and all resulting BAM, VCF, and gVCF files are included in the release.
Exploring the dataset
One can interact with the provided manifest JSON, and select for the key DRAGEN deliverables using a jq query such as below:
You could alternatively explore this dataset with the AWS CLI or via your AWS Management Console.
- From the AWS CLI
- If you do not have the AWS CLI set up yet, follow these instructions.
- Once you have the AWS CLI downloaded, you can list bucket contents using the ls command:
Repeat the ls command to explore the contents of each S3 folder.
- From the AWS Management Console
- If you are more comfortable in the AWS Management Console, just sign into the Console and navigate to https://s3.console.aws.amazon.com/s3/buckets/1000genomes-dragen
Ready to get started? Explore the DRAGEN-1kGP dataset on the Registry of Open Data—and check it regularly for new tutorials and notebooks on how to apply these data in your own work. Finally, stay tuned for our next blog post, a deeper technical dive into to the DRAGEN platform.
—
 Bryan Lajoie is a Staff Bioinformatics Scientist at Illumina Inc. in the Product Development / Customer Collaboration & Innovation (CCI) organization. Bryan and the CCI team are currently working with a diverse set of research and clinical customers to build innovative, flexible, and scalable solutions using the power of the cloud. He works across a range of Next Generation Sequencing (NGS) applications, with a focus on clinical whole genome sequencing for Rare Undiagnosed Genetic Disorders (RUGD) and the Illumina DRAGEN Bio-IT platform, which provides accurate, ultra-rapid genomic analysis of sequencing data. He received his Ph.D. from the University of Massachusetts Medical School, under the direction of Job Dekker Ph.D. HHMI, where he worked to develop and pioneer the Hi-C methodology.
Bryan Lajoie is a Staff Bioinformatics Scientist at Illumina Inc. in the Product Development / Customer Collaboration & Innovation (CCI) organization. Bryan and the CCI team are currently working with a diverse set of research and clinical customers to build innovative, flexible, and scalable solutions using the power of the cloud. He works across a range of Next Generation Sequencing (NGS) applications, with a focus on clinical whole genome sequencing for Rare Undiagnosed Genetic Disorders (RUGD) and the Illumina DRAGEN Bio-IT platform, which provides accurate, ultra-rapid genomic analysis of sequencing data. He received his Ph.D. from the University of Massachusetts Medical School, under the direction of Job Dekker Ph.D. HHMI, where he worked to develop and pioneer the Hi-C methodology.
Acknowledgements:
The authors would like to acknowledge Erin Chu, Aaron Friedman, Sapan Anand, Chris Kunard, Michael Eberle, Xiao Chen, Daniel Brami, Heidi Norton, Jay Patel, Shyamal Mehtalia, Alison Selle, Ali Crawford, Egor Dolzhenko, Andrew Gross and Rami Mehio for comments and review of the content. Sidney Kuo, Gordon Bean, Sharon Hasson for DRAGEN/Platform/Workflow support. Gordon Bean and Andrew Gross for python-notebook support. Anthony Cox, Zhuoyi Huang, Sorina Maciuca, Ole Schulz-Trieglaff and Lilian Janin for data aggregation support. The Illumina IAP and Cloud Infrastructure teams, especially Jessica Gordon, Richard Theige, Vijay Thirunavukkarasu, Keith Baylor, Charles Aylward, Ganesha Bhaskara and Jason Alexandrea. And the entire Illumina DRAGEN Software and Informatics team, especially Cobus De Beer, Anthony Cox, Severine Catreux, Shyamal Mehtalia and Rami Mehio.