AWS for Industries
Evaluating SARS-CoV-2 binding affinities using Yasara simulations on AWS
Blog guest authored by Dr. Vedat Durmaz of Innophore
In 2021, Innophore, a structural bioinformatics startup company aiming at computational enzyme and drug discovery, started the virus.watch project in cooperation with the AWS Diagnostic Development Initiative. The principal goal of this project is the implementation of a monitoring and evaluation system for emerging drug and disease-relevant Coronavirus (SARS-CoV-2) variants.
In this blog we’re going to walk you through utilizing Yasara molecular dynamics simulations on Amazon Web Services (AWS) Cloud services to quickly scale any computational demand without having to acquire costly in-house infrastructure. AWS’ on-demand capabilities are available within minutes, as soon as new SARS-CoV-2 genomes have been identified and published. Since Autumn 2021, these simulation studies have been used for two parallel strategies:
- direct high-precision binding affinity predictions for newly emerging variants and
- for the development of point cloud-based AI models as a cost-efficient alternative to molecular simulations.
Overview
Since its outbreak in Wuhan at the end of 2019, about 300 million people have been infected by a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causing more than five million confirmed deaths within the first two years [1]. Meanwhile, mutational variants (recent examples depicted in Figure 1) of the Wuhan wild-type have emerged that are characterized by higher infectiousness due to immune escape mutations and/or increased receptor binding. It is therefore a matter of public, as well as global, concern to identify highly infectious SARS-CoV-2 variants at the earliest possible moment for the sake of immediate treatment, potentially before their actual outbreak.
SARS-CoV-2 attaches to human angiotensin-converting enzyme 2 (hACE2) proteins located on the surface of various cell types via the receptor binding domain (RBD) of its spike proteins, in order to release its genome into the human host cell. According to crystallographic X-ray data retrieved from the Protein Data Bank, on the interface level, multiple viral amino acids bind to ACE2 with high precision and with an affinity (dissociation constant KD) in the order of a nanomole. Since elevated spike-receptor binding affinities generally correlate with increased infectiousness, we are targeting an early identification of SARS-CoV-2 variants associated with high affinities using computational methods.
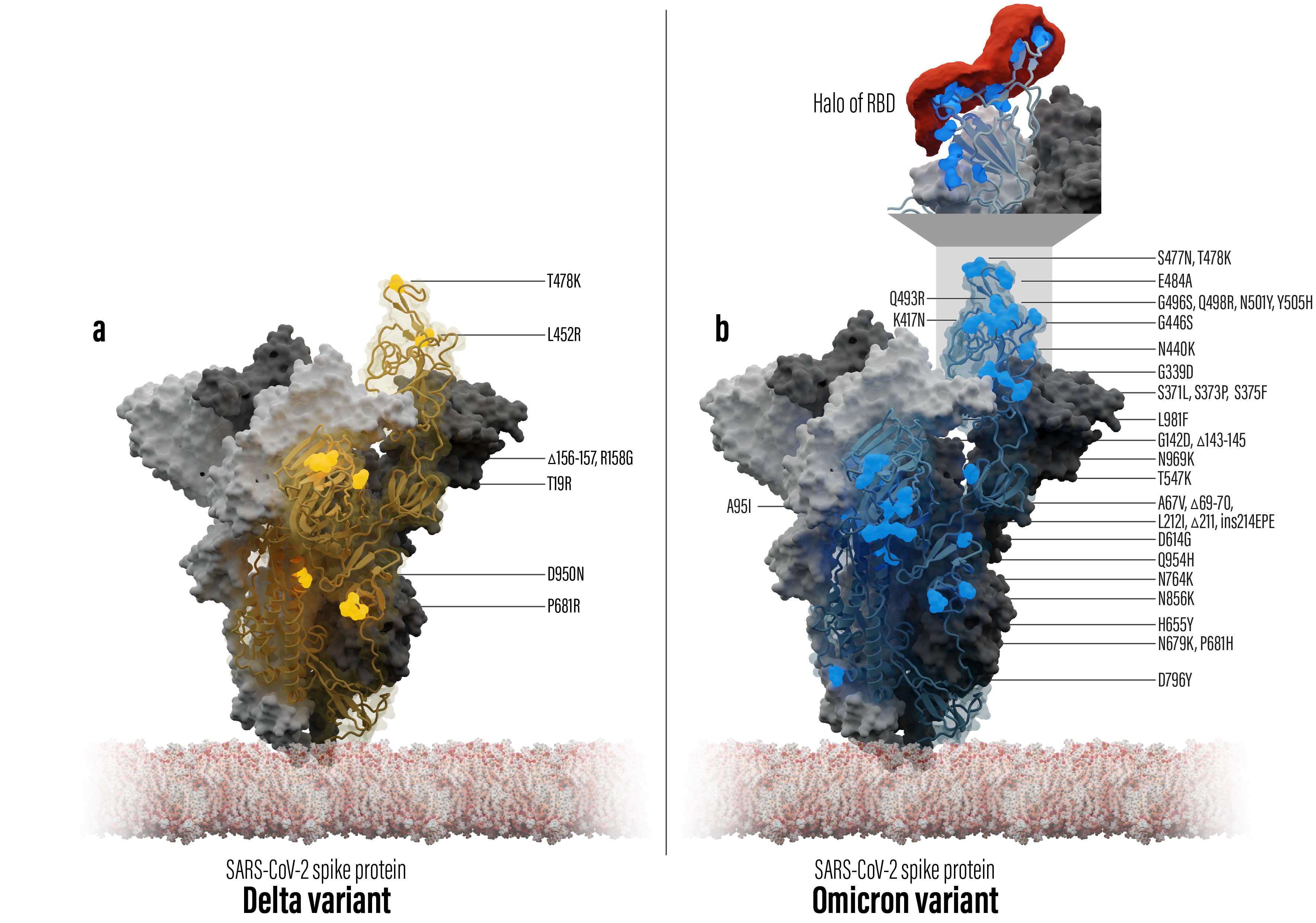 Figure 1: Spike trimer protein models of SARS-CoV-2 Delta (left) and Omicron (right) including mutation locations (shown as spheres) in comparison to the Wuhan wild type reference. One of the trimer chains is shown as a transparent surface covering the protein backbone. The top box on the Omicron structure zooms into the RBD binding interface covered by a chemical property point cloud (red) calculated with the in-house Halo point cloud toolbox.
Figure 1: Spike trimer protein models of SARS-CoV-2 Delta (left) and Omicron (right) including mutation locations (shown as spheres) in comparison to the Wuhan wild type reference. One of the trimer chains is shown as a transparent surface covering the protein backbone. The top box on the Omicron structure zooms into the RBD binding interface covered by a chemical property point cloud (red) calculated with the in-house Halo point cloud toolbox.
Atomistic molecular dynamics (MD) simulations grant access to a variety of mechanistic and thermodynamic insights into complex biological molecular systems. In particular, many macroscopic properties can be derived from microscopic snapshots sampled in accordance with the underlying dynamics. Typical applications include the estimation of binding free energies for biological complexes. However, the high predictive accuracy in binding affinities MD simulations are known for is adherent to a substantial computational demand requiring large high-performance computing or cloud computing facilities.
The virus.watch monitoring system is a derivative of Innophore’s containerized structural bioinformatics platform denoted as Catalophore and used as an internet browser-based web application. MD simulations performed in the Catalophore platform make use of the proprietary software Yasara with, in contrast to other MD engines, highly automated AMBER force field parameterization routines. Specifically, not a single manual step is required upon parameter assignment, neither for proteins nor for small molecules including substrates and cofactors.
Predictive binding energy model development
From the various MD based approaches, the empirical linear interaction energy (LIE) method developed in the 90s by Aqvist et al. [2] has proven an outstanding trade-off between predictive accuracy and computational effort. In contrast to complex methods mimicking thermodynamic reaction paths associated with high degrees of decomposition and large numbers of long trajectories, it exclusively relies on short simulations of one bound (associated) and one unbound (dissociated) state of the ligand as depicted by Figure 2. In terms of the spike-ACE2 reaction where we are dealing with two proteins rather than a protein-ligand system, we arbitrarily considered ACE2 as a ligand.
 Figure 2: Gibbs binding free energy of the spike RBD-ACE2 association/dissociation reaction.
Figure 2: Gibbs binding free energy of the spike RBD-ACE2 association/dissociation reaction.
Average ligand interaction energies with its surrounding atoms based on Van der Waals (Evdw) and electrostatic (Eelec) forces are calculated for the bound as well as unbound case. The weighted sum of Evdw and Eelec differences (bound minus unbound) finally yields Gibbs free energies of binding, ΔG.
![]()
ΔG in turn has a logarithmic relationship with the dissociation constant KD typically measured using in-vitro/in-vivo lab methods. It must be noted, though, that due to the huge ACE2 “ligand” size and, in order to reduce the impact of noise, only interactions of ACE2 amino acids in a 5 Å vicinity of spike RBD were evaluated upon interaction energy computation.
For the LIE model development, we employed a training set of 43 mostly artificial variants (mutants) of Wuhan type SARS-CoV-2 spike receptor binding domain (RBD) in complex with the human ACE2 receptor. Their structure was predicted through homology modeling using a corresponding X-ray crystal structure deposited at the Protein Data Bank (PDB) under the ID 6M0J. After 50 replicates of 500 ps MD simulations we observed a sufficient cumulative average binding energy convergence characterized by a negligible number of rank swaps (comparing replicate on replicate) between every pair of two variants as illustrated by the green plot in Figure 3. One could, however, accelerate things if using 30 replicates, but at the cost of less accuracy. In contrast, the distribution’s overall average and standard deviation converge much earlier.
 Figure 3: Cumulative average binding energy convergence of 43 training set variants with increasing number of MD replicates. Blue plot: mean binding energy with standard deviation. Green plot: fraction of variant pairs associated with rank swaps.
Figure 3: Cumulative average binding energy convergence of 43 training set variants with increasing number of MD replicates. Blue plot: mean binding energy with standard deviation. Green plot: fraction of variant pairs associated with rank swaps.
Using linear regression, interaction energy weights of the empirical model were fitted to ΔG values derived from KD values, which had been measured and recently published by Zahradnik et al. [3] With our final binding energy model
![]()
(available at Nature Scientific Reports [4]) we achieved squared coefficients of correlation R2 amounting to 0.79 and, respectively, 0.71 for fitting and leave-one-out cross-validation and mean absolute errors of only 2.0 and 2.1 kJ/mol. This is less than 4% of the average predicted, as well as lab binding affinity (green and orange scatter plots in Figure 4). Moreover, for three spike variants of concern (VOC), namely Alpha, Beta, Gamma and, in addition the Wuhan wild-type (blue markers), the order of predicted binding energy is in agreement with the experimentally determined affinities. Regarding the complexity of protein-protein binding energy estimations, this model’s accuracy is more than satisfactory. We will consequently use it on AWS for monitoring newly emerging SARS-CoV-2 variants.
 Figure 4: Scatter plot of fitted (orange) and cross-validated (LOO, green) vs. experimental spike RBD-ACE2 binding energies along with coefficients of determination and mean absolute errors. Application to VOC (blue squares).
Figure 4: Scatter plot of fitted (orange) and cross-validated (LOO, green) vs. experimental spike RBD-ACE2 binding energies along with coefficients of determination and mean absolute errors. Application to VOC (blue squares).
Performance evaluation of MD simulation on Amazon EC2 compute optimized instances
For the performance evaluation on Amazon Elastic Compute Cloud (Amazon EC2) instances of different instance types, we used the SARS-CoV-2 Omicron spike RBD in complex with hACE2. It was placed in a cuboid simulation box with a 5 Å distance to the complex on each side yielding roughly 85,000 atoms after explicit solvation with TIP3P.
Only Amazon EC2 instances of the compute optimized instance class were considered. We consequently focused on Intel-based architectures of the C5 and C6i classes, thereby investigating scalability with respect to CPU core numbers. Instances of the C5 and C6i class were launched using the Amazon Linux 2 Kernel 5.10 AMI. On each instance, this initial molecular system underwent 50 independent 500 ps MD simulations resulting in 50 trajectory replicates with a total simulation time of 25 ns. On each EC2 instance, one Docker container was launched that sequentially produced all the 50 MD replicates.
We compared each type of the C5 and C6i instance classes. Table 1 summarizes run time and costs per SARS-CoV-2 variant according to the North Virginia region (us-east-1), performance measures including the simulation time achieved per day, average CPU usage and, finally, predicted binding affinities.
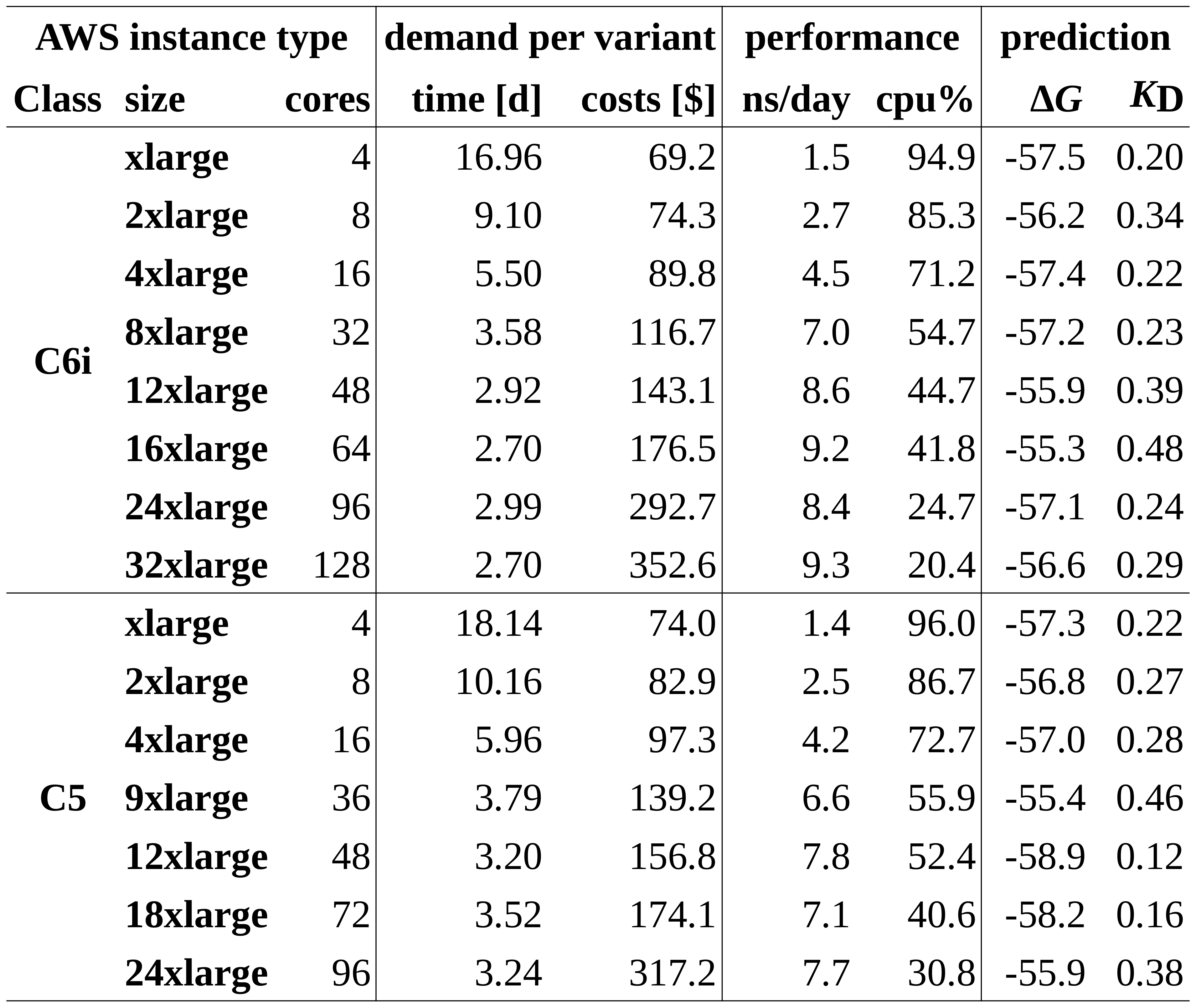 Table 1: Demand per SARS-CoV-2 and performance of C6i and C5 instances
Table 1: Demand per SARS-CoV-2 and performance of C6i and C5 instances
 Figure 5: Run time and costs per SARS-CoV-2 variant along with MD simulation performance versus number of CPU cores of Amazon EC2 C6i and C5 instances.
Figure 5: Run time and costs per SARS-CoV-2 variant along with MD simulation performance versus number of CPU cores of Amazon EC2 C6i and C5 instances.
With an average energy of -56.98 kJ/mol and a standard deviation of 1.1 kJ/mol, all instances yield, as expected, comparable final binding affinities within a tight range. However, the huge differences in terms of performance and costs are better visualized in a graphical plot as shown in Figure 5.
As a first observation, C6i instances (blue solid line) perform about 3-10% faster than C5 instances (blue dashed line) when comparing instances with the same number of cores. In case of both instance classes C5 and C6i, the run time (green plots) cannot be significantly reduced by using more than 48 cores for the given simulation box consisting of 85k atoms. Whereas the costs per virus variant (orange lines) further substantially increase.
The 14% and 18% performance boost with 48 compared to 32 cores (C6i) and, respectively, 36 cores (C5) cores seems to be the maximum reasonable gain that can be considered in order to accelerate simulations. This is considerably less than the 57-58% performance boosts still achieved when switching from 16 to 32/36 cores. With another boost of 7.5% compared to 48 core nodes of the C6i class, C6i.16xlarge instances with 64 cores performed best (together with 128 core C6i.16xlarge instances) and thus can be considered in very urgent situations. However, the minimal improvement comes at 23% higher costs.
Conclusion
According to our performance results, which are specific to Yasara MD simulations of nearly 100k atom systems, the optimal instance setup in case of particularly urgent investigations would comprise the 64 core C6i.16xlarge instance. However, for less urgent studies, we will most likely utilize the C6i.8xlarge or even C6i.4xlarge instance with 32 and, respectively, 16 cores.
Upon model development and optimization, each run of the entire 43 item training set required 43 either C6i.4xlarge or C6i.8xlarge instances in order to fit the two model parameters. This amounted to nearly 700 or 1400 cores that were immediately available for run times of 3-5 days. Compared to our in-house computing facilities with less and outdated nodes, where these calculations would have taken several weeks, we were able to reduce the computing time to less than 20% and thereby save more than 30% of power costs.
For these and other reasons including high virtualization and containerization levels, as well as a broad range of hardware architectures, the AWS Cloud scalable computing infrastructure is well suited for computationally demanding scientific studies.
The AWS Management Console provides many useful tools for project management and control over hardware and docker containers. With the aid of browser-based monitoring tools and the Amazon EC2 serial console, the calculations and hardware performance of running instances are easily overviewed. Thanks to the AWS Cost Explorer, we are able to keep an eye on the costs that can be differentiated between instance types and appropriately plotted over variable time ranges.
Getting started with Amazon Web Services
References
[1] WHO. Coronavirus disease (COVID-19) pandemic, https://www.who.int/emergencies/diseases/novel-coronavirus-2019. (2021).
[2] Aqvist, J., Medina, C. & Samuelsson, J. E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 7, 385–391 (1994).
[3] Zahradník, J. et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 6, 1188–1198 (2021).
[4] Durmaz, V., Köchl, K., Krassnigg, A. et al. Structural bioinformatics analysis of SARS-CoV-2 variants reveals higher hACE2 receptor binding affinity for Omicron B.1.1.529 spike RBD compared to wild type reference. Sci. Rep. 12, 14534 (2022).
DOI: https://doi.org/10.1038/s41598-022-18507-y
 Dr. Vedat Durmaz is Head of Computational Chemistry and Data Engineering at the Austrian startup company Innophore. His responsibilities comprise software development for molecular dynamics and bioinformatics workflows as well as related data science and virtual containerization on HPC/cloud computing facilities. Vedat has a Diploma Engineer degree in Biotechnology as well as a Master of Science and Ph. D. degree in Bioinformatics from the Freie Universität Berlin in cooperation with the numerical mathematics department of the Zuse Institute Berlin. Since 2019, he supports Innophore in enzyme and drug discovery as well as protein engineering.
Dr. Vedat Durmaz is Head of Computational Chemistry and Data Engineering at the Austrian startup company Innophore. His responsibilities comprise software development for molecular dynamics and bioinformatics workflows as well as related data science and virtual containerization on HPC/cloud computing facilities. Vedat has a Diploma Engineer degree in Biotechnology as well as a Master of Science and Ph. D. degree in Bioinformatics from the Freie Universität Berlin in cooperation with the numerical mathematics department of the Zuse Institute Berlin. Since 2019, he supports Innophore in enzyme and drug discovery as well as protein engineering.