AWS Startups Blog
How Stoke Therapeutics Is Turbo-Charging Drug Discovery Using Snakemake and AWS
By Eric Lim, Head of Bioinformatics at Stoke Therapeutics
As a biotechnology company, Stoke Therapeutics is dedicated to addressing the underlying cause of severe diseases by upregulating protein expression with RNA-based medicines. Identifying genomic signatures amendable to TANGO (Targeted Augmentation of Nuclear Gene Output), our proprietary approach to developing antisense oligonucleotides and selectively restore protein levels begins with computational analyses of both publicly-available archived data and privately-generated sequencing data. Through AWS, Stoke has quick access to computing resources without the active management of on-prem hardware, enabling us to close the gap between sequencing and interpretation and dedicate more time to science. Our proprietary bioinformatics pipelines have identified approximately 2,900 genetic diseases, which we believe to be amenable to TANGO.
Today, we’re sharing how we leverage AWS services, some scalability constraints we experienced in our journey, and the architecture that has worked well for us. To date, we have deployed this cloud-native infrastructure to analyze over 20,000 RNA sequencing samples with an estimated cost saving of over 70% when compared to a traditional approach using on-demand Amazon EC2 VMs with local EBS storage.
Overview of architectural design in AWS
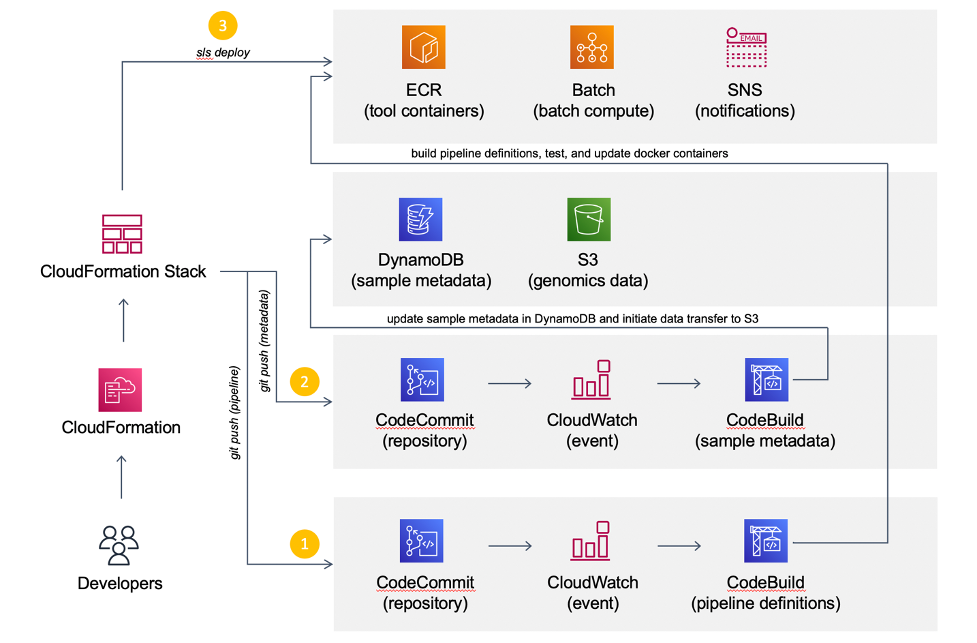
CI/CD and workflow management in AWS CodeBuild
Our first program addressing Dravet syndrome, a severe and progressive genetic epilepsy, is in clinical settings. We are also pursuing a second haploinsufficient disease, autosomal dominant optic atrophy (ADOA), the most common inherited optic nerve disorder. Our bioinformatics workflows in analyzing genomics data are composed of several disjointed command-line utility tools that transform raw sequencing data into interpretable genomic signatures to identify the relevant disease and drug targets. Different tools often have their own set of inputs, outputs, resource footprints, and software requirements, so we use a workflow management system to manage versioning and dependencies between jobs, and most importantly, to ensure in silico reproducibility. At Stoke, we use Snakemake and conda/mamba to deploy analytical workflows within the AWS ecosystem of services. For more information, you can also review an incomplete but long list of existing workflow management systems used in bioinformatics.
Continuous Integration
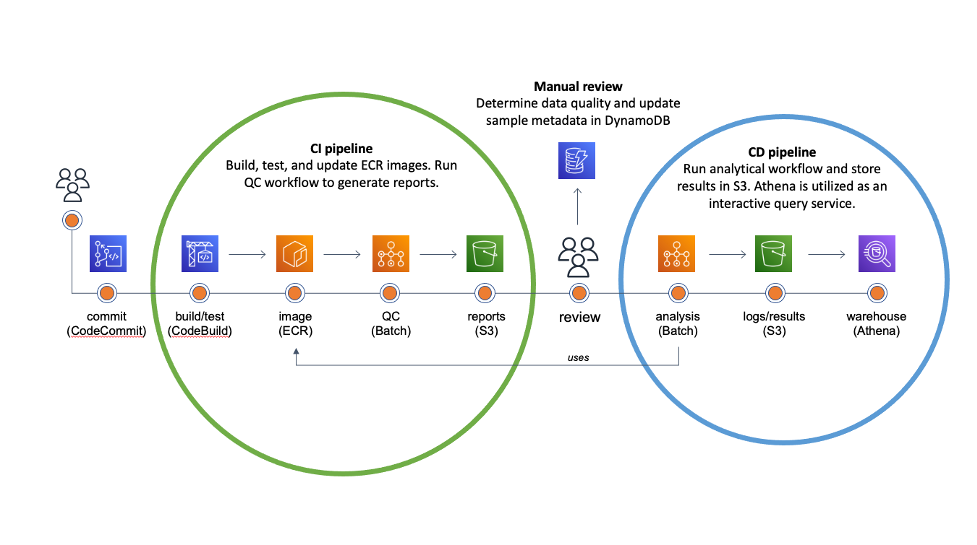
Continuous Integration/Continuous Deployment (or Delivery) is the backbone of modern software and DevOps environments. Our goal is to transform hundreds of terabytes of genomics data into queryable scientific insights by automating as much as possible the deployment of our analytical workflows. The diagram below illustrates our approach.
We use CodeCommit as our Git repository. The team also subscribes to trunk-based development, where developers collaborate on a single branch, to avoid long-running branches from falling out of sync. Consistent with others, this methodology has substantially reduced many frustrating scenarios with merging code.
Once the code is committed and pushed into the master branch, AWS CodeBuild takes over to create a Docker image containing the pipeline definitions and install software requirements in the specified versions. Snakemake natively supports automated conda deployment via mamba (–conda-create-envs-only and –conda-frontend mamba) and a modest unit testing framework (–generate-unit-tests), increasing reproducibility. CodeBuild pushes the resulting image to the corresponding Amazon ECR registry that EC2 instances provisioned from AWS Batch will consume. Finally, we incorporate the quality control (QC) workflow as part of the CI pipeline to automatically assess data quality and store QC reports in S3.
Review
The majority of our sequencing data comes from the scientific literature. The reuse of publicly accessible data has immense potential for novel scientific discoveries, but there are often inherent quality differences between data sources. While deployment can be fully automated, we deemed that the data quality assessment is an important business decision. So, the team manually reviews QC reports for each genomic project and updates the corresponding metadata in Amazon DyanmoDB before triggering analytical deployments.
Continuous Delivery
The CI pipeline uses CodeBuild to automatically generate all necessary ECR images for the CD pipeline to deploy the analytical workflow to Batch. The workflow stores all intermediate data, essential log files, and results in S3. Results are converted into Parquet and compressed with Snappy. We utilize Amazon Athena and Amazon QuickSight as an interactive query service to analyze genomic results in a data lake using standard SQL.
Metadata in AWS DynamoDB
We use DynamoDB to store metadata of genomic samples. When DynamoDB receives a new item, the stream triggers a Lambda function to create a Batch job, which downloads the corresponding raw sequencing data from public repositories such as the Gene Expression Omnibus to S3.
Compute strategy in AWS Batch
Our compute approach is straightforward and predominantly uses AWS Batch to process genomics data. Batch is a fully managed service that enables us to scale our containerized genomic workflows to hundreds of thousands of compute nodes, all without actively managing the infrastructure. The diagram below illustrates our strategy.
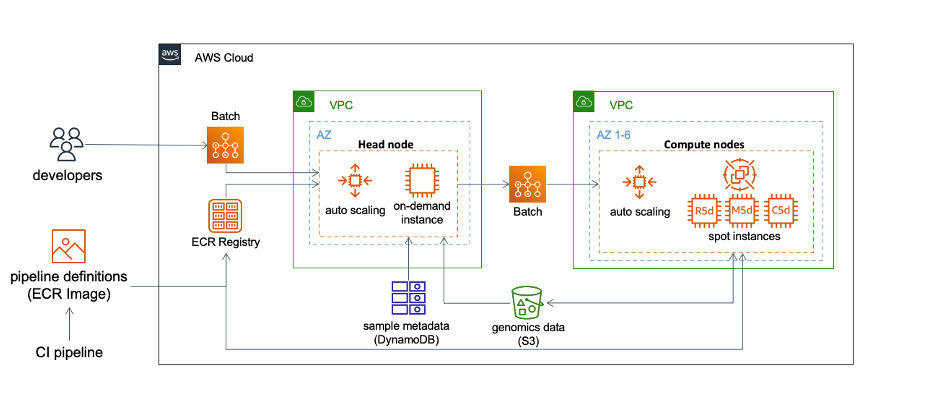
A workflow may contain many independent container jobs. As each job may or may not run on the same instance, Snakemake offers several utility functions to facilitate the deployment.
We use S3 as a central storage service to exchange genomics data, results, and logs between containers. Snakemake natively supports remote I/O (–default-remote-provider S3 and –default-remote-prefix bucket_name). In addition, the functions also handle staging remote data before job execution, uploading outputs to S3 as job completes, and performing cleanup to free up disk space for jobs remaining in the queue.
We deploy the entire Snakemake workflow to Batch, which launches an on-demand EC2 instance serving as a head node to manage job dependencies and submission. Workloads defined in the Batch job queue autoscale the provision of Spot instances. The head node will run for the entire duration of the workflow, whereas each Spot instance will terminate when it is no longer processing jobs. We predominantly use 3-5 EC2 Spot instance types that provide an instance store, including R5d, M5d, and C5d. An instance store provides temporary block-level storage that is physically attached to the instance, and in our experience, produces superior I/O performance for genomics analyses. This approach eliminates using EBS as the compute storage, which further reduces costs.
Scalability
As a result, we have deployed this strategy to analyze over 20,000 RNA sequencing samples in a cost-effective and performant manner. However, as data grow exponentially and analyses inherently become more complex, we started hitting a few scalability constraints. Although entirely relying on instance store as the compute storage is both cost-saving and performant, the size of the instance store might not always offer enough capacity to accommodate every job. Without carefully planning for vCPU/GPU, memory, and storage requirements, jobs occasionally fail from running out of disk space.
Amazon FSx for Lustre provides transparent synchronization of data stored in S3 and is accessible as a shared filesystem across all instances. The service is attractive as most bioinformatics tools natively work with data stored on a POSIX-compliant filesystem. The team is currently evaluating FSx for Lustre as an alternative solution for genomic workflows that require large amounts of disk space per job.
Our strategy relies on S3 as a central storage location. Building job dependencies with many remote input and output I/O objects can be a time-consuming task. Some of our workflows take over several hours of processing before Batch deployment.
We implemented a custom cluster execution logic in Snakemake (–jobscript) to alleviate this constraint. When deploying a workflow, we instruct Snakemake to always run every job without first checking if the outputs already exist in S3. It allows us to delay interacting with remote I/O objects until the actual submission to Batch, a step we can parallelize and scale with ease. If the expected output of a job is already in S3, we interrupt Snakemake from submitting the job to Batch and record the job as complete. Instead of spending hours preprocessing all job dependencies of large workflows, this approach takes just minutes to start sending unprocessed jobs to the Batch queue.
Conclusion
Although Stoke is now a publicly traded and clinical-stage biotech company, our AWS journey began when we had just a few employees. AWS enables startups like us to stay agile and scale the compute infrastructure as we grow. Growth does come with pains, but we are continually improving and leveraging the latest technology in AWS to further our understanding of genetic diseases.
 |
Eric Lim received his Ph.D. from Brown University in 2011. His work focused on using computational biology and high throughput genomics techniques to identify functional splicing elements in the genome. Eric has a decade of postdoctoral experience in the biotech industry and is currently the Head of Bioinformatics at Stoke. He leads a team of talented computational scientists in leveraging antisense oligonucleotides as a therapeutic modality to treat the underlying cause of genetic diseases. He has over 24 publications and patents in RNA splicing, and his work has led to numerous awards, including the 40 Under 40 in Cancer. Eric is also a member of the COVID-19 International Research Team. Acknowledgements The bioinformatics and IT teams have contributed substantially to the success in using AWS to process genomics data. I want to take this opportunity to acknowledge Sebastien Weyn, Jose Negron, and Stephen Yu. |