AWS Quantum Technologies Blog
Classiq and AWS Power Quantum-Classical Chemistry Innovation in Singapore with Hatch
Introduction
In biochemical processes development and analysis, binding energy, the energy released when a small molecule docks into a protein’s active site, determines how strongly a compound, such as a ligand, interacts with its protein target. Early-stage computational prediction of this quantity helps research teams prioritize candidates before committing to resource-intensive laboratory testing. Conventional methods in chemistry handle this well for small systems, but face known accuracy limitations in strongly correlated molecular environments and incur growing computational costs as system size increases. Addressing both challenges is the motivation for this work.
This post describes a project by Classiq to build and validate a complete quantum-classical pipeline. The project was conducted as part of Dimension X, an open innovation challenge by Hatch, an innovation center in Singapore. The goal of the project was to demonstrate a quantum-classical pipeline for computational chemistry and binding energy estimation. The calculations were conducted on AWS cloud infrastructure using Amazon Elastic Compute Cloud (Amazon EC2) on a c6i.16xlarge instance with 64 vCPUs and 128 GB of memory. The pipeline combines high-performance parallelized Density Functional Theory (DFT) calculations with a variational quantum eigensolver (VQE), made accessible through the Classiq platform. The result is a workflow that uses AWS resources to handle the heavy classical computation and quantum computing to account for quantum correlations in the calculations, increasing accuracy beyond what DFT alone provides.
What you will learn from this post:
- How ligand-protein interactions and binding energy estimation are formulated as a quantum chemistry problem
- How Classiq built and ran a hybrid classical–quantum workflow using AWS compute infrastructure.
- How the fragment-environment embedding approach keeps the quantum problem tractable as system size grows
The Problem: Why Binding Energy Prediction Is Hard, and Why Quantum Helps
A central mechanism in many biochemical processes is molecular recognition: a small molecule, or ligand, binds to a specific site on a larger biomolecule, usually a protein, and changes its activity. This interaction is fundamental across applications such as enzyme modulation, molecular screening, target engagement analysis, and biochemical process optimization. This same binding mechanism also underlies the therapeutic effects of many medicines; a small molecule binds to a target protein, modulating its activity to produce a desired biological effect.
The region of the protein where the ligand binds is called the binding pocket or active site. A key step in analyzing ligand–protein interactions is estimating the compound’s binding affinity for relevant biological targets and candidate binding pockets. Stronger binding energy generally indicates tighter ligand–protein binding at the active site, which can affect how strongly the molecule modulates the target’s function.
Computationally, this reduces to:
ΔE = E(complex) − E(pocket) − E(ligand)
Evaluating the binding energy, therefore, requires three separate quantum chemistry calculations. Each calculation is performed on a system that, in a realistic target, can contain 100+ atoms and hundreds of interacting electrons, even when modeling only the local binding pocket.
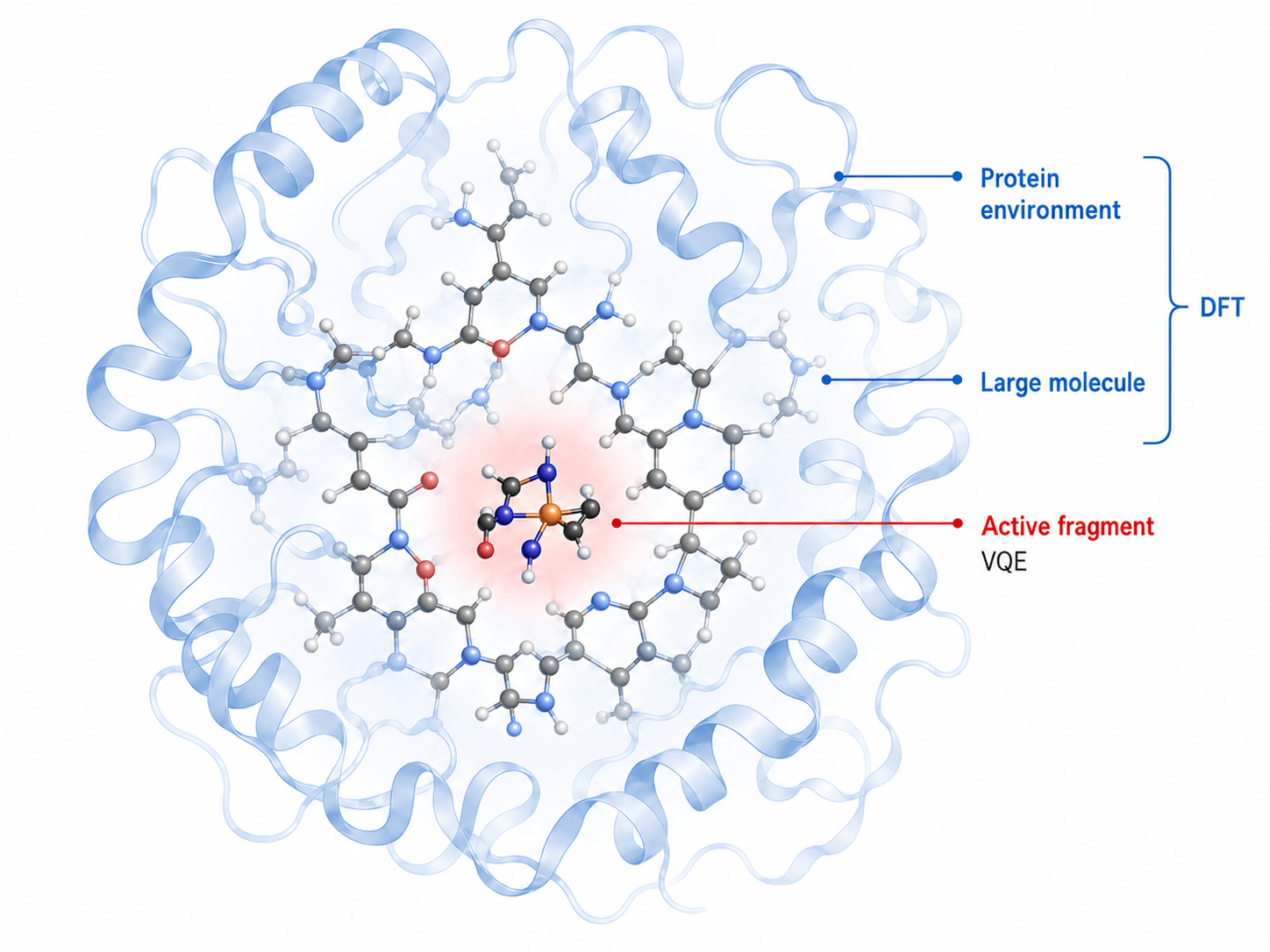
Figure 1: Conceptual schematic representation of a ligand-protein binding pocket. The fragment region (active space) is treated quantum mechanically via VQE, while the surrounding protein environment is handled with DFT. This partition keeps the quantum problem tractable regardless of the full system size. The figure is illustrative of the computational partitioning used in the workflow.
Classical approaches face a fundamental trade-off between computational cost and physical accuracy. Molecular mechanics methods such as Molecular Mechanics Generalized Born Surface Area (MM-GBSA) are computationally efficient, but they rely on parameterized classical force fields and do not explicitly model the electronic structure. As a result, quantum effects are captured only indirectly through force-field parameterization and approximations. DFT offers a better balance: it scales polynomially with system size (number of electrons) and provides accurate results for many molecular systems. However, it still has known limitations for noncovalent interactions, charge transfer, dispersion, transition-metal centers, and strongly correlated electronic structures. Wavefunction methods such as Full Configuration Interaction (FCI) are formally exact within a chosen basis, but scale exponentially with system size, making them infeasible for realistic molecules.
Quantum computing offers a potential path through this bottleneck. In this proof-of-concept, we use a standard VQE to demonstrate the feasibility of the hybrid quantum-classical approach. While this initial implementation deliberately uses standard VQE, the architecture is modular by design and can support more advanced quantum algorithms as hardware and methods mature. In the noisy intermediate-scale quantum (NISQ) regime, this includes VQE-based methods for quantum chemistry (Rossmannek et al., 2023 [1]), which have shown promising resource-efficient performance for strongly correlated fragments. In the fault-tolerant regime, the same framework could be extended toward algorithms such as Quantum Phase Estimation (QPE), enabling higher-precision energy estimation. The present work should be viewed as a proof-of-concept step toward future quantum-enhanced evaluation of binding energies in chemically active sites, where high-accuracy classical treatments can become computationally expensive.
The challenge is that current quantum hardware has limited qubit counts, so the quantum calculation must target a small, carefully chosen subset of the molecule. The subset of atoms defines the active space where the most important electronic correlations occur. Even after the active space is chosen, researchers still face the circuit-engineering challenge of translating the quantum chemistry problem into an efficient circuit that fits hardware constraints. The Classiq platform addresses the challenge directly: researchers specify the quantum chemistry problem, using the Classiq SDK, and the platform synthesizes optimized quantum circuits without requiring gate-level programming. This abstraction is especially important as hardware matures. The same high-level model can be retargeted, optimized, and scaled to larger active spaces and deeper circuits, reducing a major practical barrier to experimenting with VQE today while preparing workflows for more capable quantum machines.
The Pipeline: From Crystal Structure to Binding Energy
Classiq built a pipeline that is validated progressively against molecular systems of increasing complexity. The approach is grounded in projection-based Wavefunction-in-DFT embedding (Lee et al., 2019 [2]) and its extension to quantum computers via VQE (Rossmannek et al., 2023).
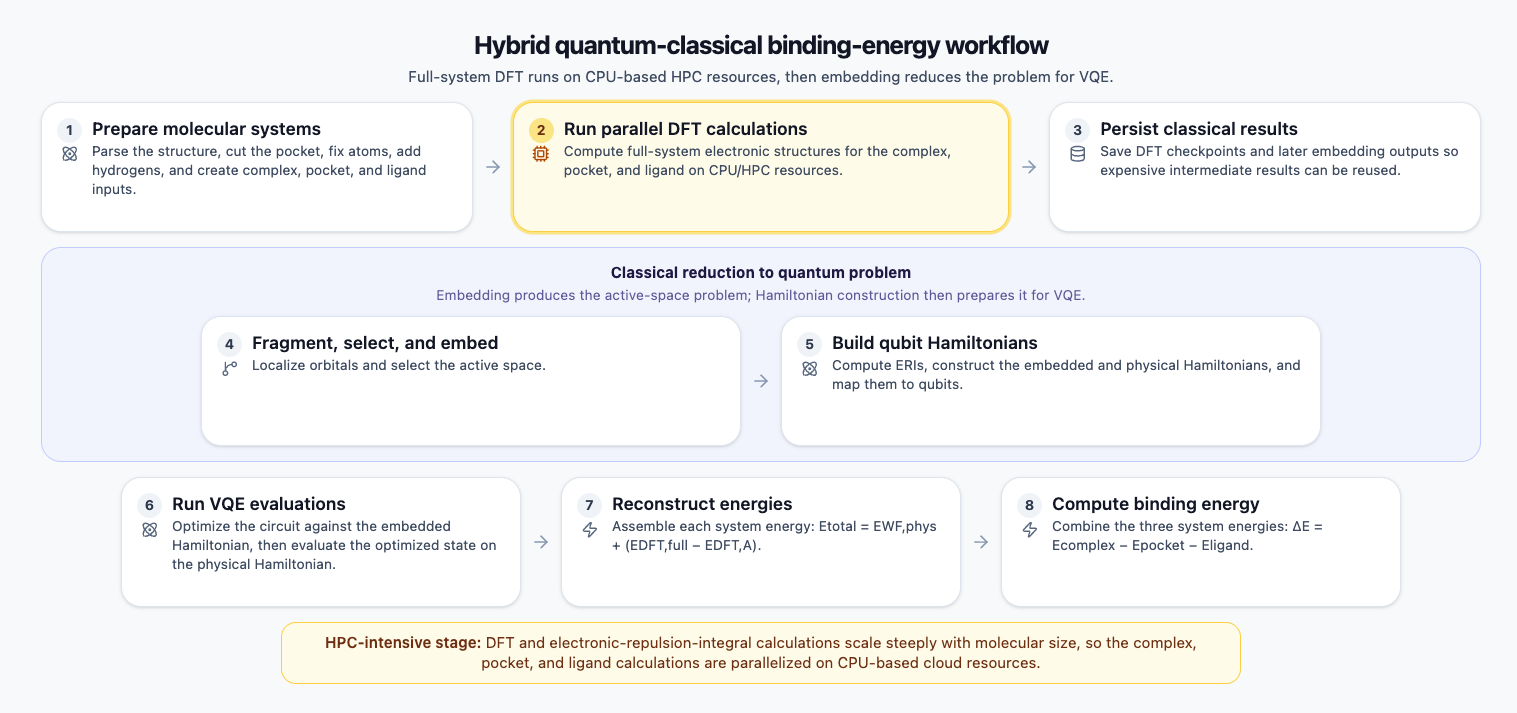
Figure 2: A hybrid quantum-classical pipeline for binding energy calculation. Classical DFT runs on HPC resources to characterize the full molecular system, embedding reduces the problem to a tractable active space, and VQE evaluates the ground-state energy of the resulting qubit Hamiltonian. The three system energies are then combined to yield the final binding energy.
The Hamiltonian embedding pipeline partitions the full molecular system into two coupled regions: a chemically active fragment and a surrounding environment. This allows the fragment, where the most important electronic correlations occur, to be treated with a quantum method such as VQE, while the larger environment is treated with DFT. The two energy contributions are then combined in a physically consistent way, improving accuracy without requiring a full quantum calculation of the entire protein system.
The first stage takes the raw crystallographic structure of the protein-ligand complex from the Protein Data Bank, cuts the structure to include the pocket only, and repairs it by adding missing atoms at the severed bonds and setting the approximate protonation state at physiological pH. A DFT calculation then characterizes the electronic structure of each of the three systems: the complex, the pocket, and the ligand separately. The results are saved to the attached Amazon Elastic Block Store (Amazon EBS) volume, decoupling this expensive first stage from downstream work.
The second stage performs projection-based Wavefunction-in-DFT (WF-in-DFT) embedding. The occupied molecular orbitals are localized and partitioned into an active space, the fragment, chemically involved in binding, and an environment comprising the rest of the protein. The environment’s electronic influence is incorporated into a modified one-electron Hamiltonian for the fragment, and the full electronic Hamiltonian (including the electron-electron interaction term) of the active space is then mapped to a qubit Hamiltonian via the Jordan-Wigner transformation. This stage is where the problem reduction happens: a system of up to ~100 atoms is reduced to an active space of roughly 10–14 spatial orbitals, with the environment’s electronic influence consistently incorporated into the fragment’s Hamiltonian, and the quantum calculation inherits the accuracy of the full-system DFT description of the environment at no extra quantum cost.
The third stage is VQE, executed through the Classiq platform. The Classiq platform allows users to build high-level functional quantum models. A synthesis engine then takes the model, along with optimization preferences and constraints (such as circuit depth, qubit count, or target hardware architecture), to produce an optimized quantum circuit without requiring manual gate-level engineering. In this workflow, Classiq’s ability to synthesize optimized circuits from high-level functional models serves as the quantum layer of the hybrid pipeline: instead of hand-engineering a circuit, the problem is described at the chemistry level, specifying the fermionic Hamiltonian, the electron counts, and the desired ansatz type. The Classiq platform then applies symmetry reduction to shrink the qubit register (qubit tapering) and constructs the Unitary Coupled Cluster (UCC) ansatz by synthesizing an optimized circuit for the hybrid execution. Execution is then handled through dedicated library functions, where the Classiq execution scheme evaluates Hamiltonian expectation values at each optimization step, allowing a standard classical optimizer to drive the VQE hybrid execution. The convergence of the VQE execution gives the final fragment energy, which is the Hamiltonian expectation. That fragment energy is then combined with the DFT environment contribution via the WF-in-DFT formula to reconstruct the total system energy, which is computed for each of the three subsystems and combined to yield the final binding energy.
The following code excerpt exemplifies the usage of the Classiq SDK to synthesize VQE circuits:
@qfunc
def main(params: CArray[CReal, num_params], state: Output[QArray]):
prepare_basis_state(hf_state, state)
multi_suzuki_trotter(uccsd_hamiltonians, params, 1, 1, state)
qprog = synthesize(main)
show(qprog)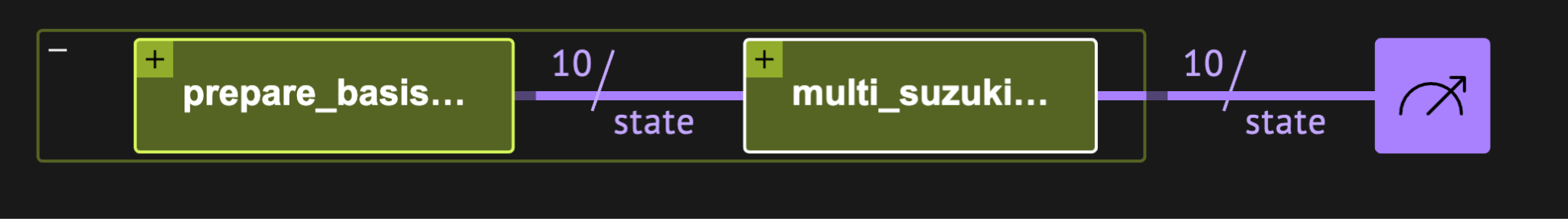
A VQE circuit as visualized in the Classiq platform. The circuit prepares a Hartree-Fock reference state and applies a Unitary Coupled Cluster (UCC) ansatz via Suzuki-Trotter decomposition, synthesized automatically from a high-level model without manual gate-level programming.
The pipeline was first validated on controlled benchmark systems. For LiH⁺, a minimal open-shell test case with a well-known exact solution, VQE-in-DFT produced a total energy of −7.620827 Ha, within 0.088% of the exact FCI reference of −7.6141565 Ha, and closer to the FCI reference than the full-DFT result of −7.663977 Ha. For H₂O, VQE-in-DFT came in at −76.205919 Ha, within 0.281% of FCI-in-DFT. In both cases, the quantum results matched or surpassed classical DFT precision. The pipeline was later validated on more complex systems, requiring the use of HPC resources, as outlined in Section 4.
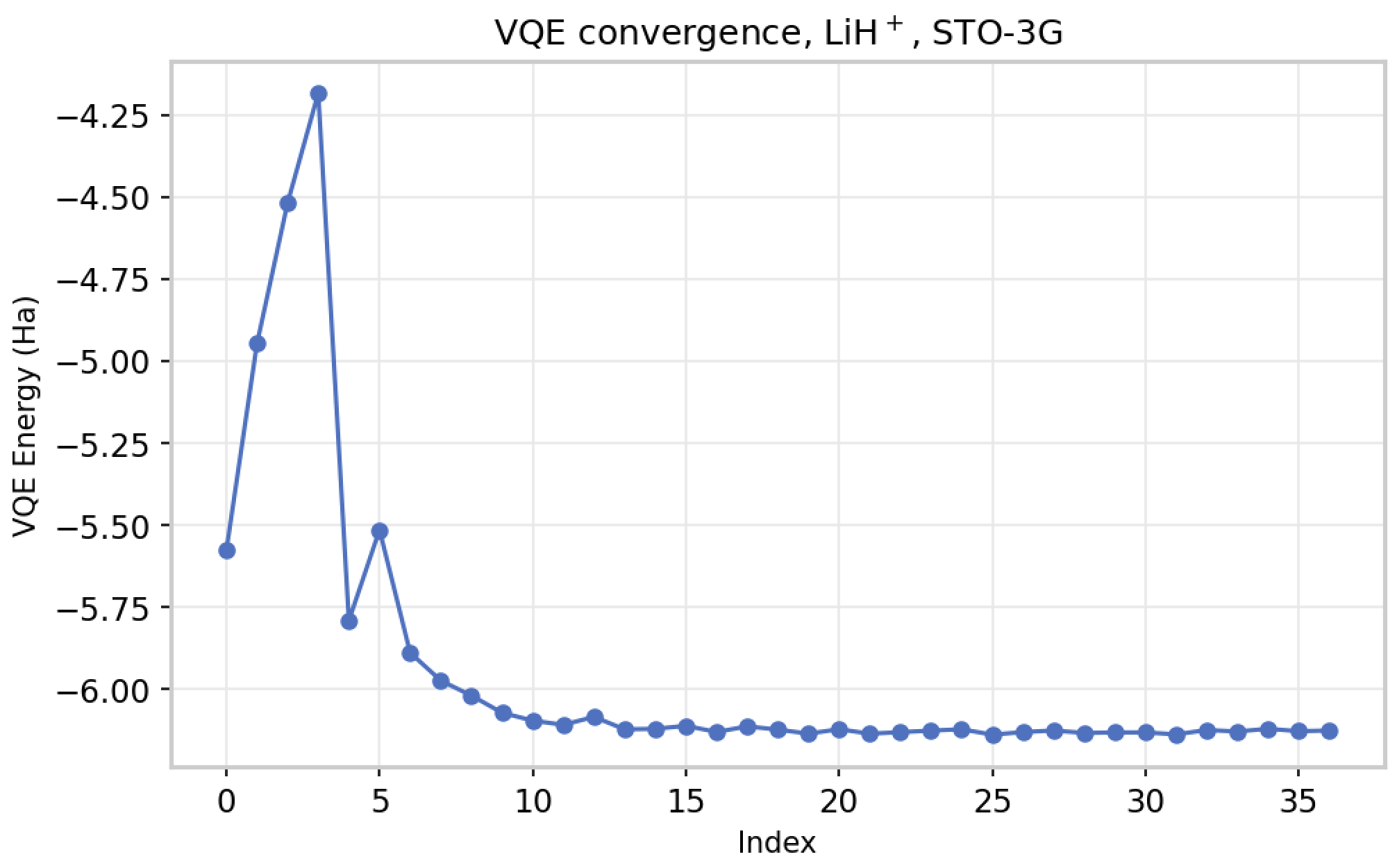
VQE energy convergence for LiH⁺ (STO-3G) as a function of optimizer iterations, using COBYLA to minimize the active-space Hamiltonian energy. The optimizer drives the UCC ansatz parameters toward the ground-state energy, which is subsequently combined with the DFT environment contribution via the WF-in-DFT formula to reconstruct the total system energy.
Technical Implementation: HPC on AWS
The Scaling Problem, and Why the Architecture Is Designed for It
Standard DFT calculations scale as O(N⁴) in the number of basis functions N (the number of functions scale linearly with the number of atoms), making them the computational bottleneck for large molecules such as enzymes.
The three DFT calculations (complex, ligand, and pocket) are computationally independent, but they do not stay the same size. Every increase in pocket radius brings more atoms into the calculation and compounds the cost: doubling the atom count multiplies the ERI computation by roughly a factor of 16. Every new ligand configuration tested against the same pocket requires re-running the complex calculation. The pipeline also exposes five control parameters: pocket radius, orbital weight threshold, number of active virtuals, and two Hamiltonian truncation thresholds, which are systematically swept during validation, further increasing the computational load. For realistic screening campaigns across multiple biochemical candidates and target conformations, this becomes a problem best solved on high-performance computers (HPC).
The fragment-environment split decouples the size of the quantum problem from the size of the classical one. As the protein pocket grows, the DFT and ERI costs scale accordingly, but the scope of the active space passed to the quantum computer is a free parameter, set independently based on the available hardware and calculation accuracy requirements. On more capable hardware, it can be expanded to capture more correlation; on more constrained hardware, it can be reduced. The classical pipeline scales with the chemistry; the quantum stage scales with the hardware.
Parallelizing DFT on Amazon EC2
To fully saturate the available hardware, the pipeline pins every major numerical backend to the full CPU count at startup. On a c6i.16xlarge (a 64 vCPUs, 128 GB memory) Amazon EC2 instance, PySCF distributes both the DFT self-consistent field (SCF) loop and the Electronic Repulsion Integrals (ERI) computation across the available cores simultaneously. These operations scale as O(N⁴) in basis size and would be intractable on a typical single-user development machine, motivating the use of a 64-vCPU Amazon EC2 instance.
The integration of the Classiq platform into the pipeline occurs at the boundary between the classical and quantum stages. Once DFT has characterized the full molecular system on the Amazon EC2 instance, and the embedding step has reduced the environment’s influence into a fragment Hamiltonian, the Classiq synthesis engine compiles the active-space Hamiltonian into an optimized VQE circuit.
In this demonstration, the VQE circuit is executed on a simulator. The same Classiq quantum model, however, can be synthesized for different execution targets, including hardware available through Amazon Braket. This means the circuit can be generated with backend-specific constraints, such as qubit connectivity, supported gate sets, and optimization preferences, rather than being hand-adapted at the gate level. In a hardware-backed deployment, the pipeline can then be routed to Amazon Braket by changing the execution backend. Braket serves as the managed execution layer for submitting the synthesized circuit to a supported QPU or simulator, collecting measurement results, and returning expectation-value estimates to the classical optimization loop. A classical optimizer iteratively updates the circuit parameters until the energy converges. The Classiq executor then handles the full parameter optimization loop internally, iterating until the energy converges without requiring explicit implementation from the user. The resulting fragment energy is passed back to the WF-in-DFT formula, where it is combined with the DFT environment contribution to reconstruct the total system energy. This is repeated for the complex, the pocket, and the ligand separately, and finally combined to yield the binding energy.
AWS Batch for Parallel Campaigns
Since the three system calculations are fully independent, they map directly onto AWS Batch, a managed service for running batch computing workloads that automatically schedules jobs, provisions the required compute resources, and scales execution across Amazon EC2 instances based on workload demand. With AWS Batch, each calculation can run as a separate job on its own Amazon EC2 instance, with different CPU and memory specifications. Checkpoints are written to Amazon Simple Storage Service (Amazon S3) and downstream embedding jobs triggered automatically on arrival. Since the pipeline is parallelized, the total runtime is equal to the slowest single job, not the sum of the three.
The same architecture extends to screening campaigns, in which dozens or hundreds of ligand configurations are tested against the same pocket. Each configuration becomes an independent Batch job, with cost scaling linearly. With this design, many ligand configurations can be evaluated in parallel, while AWS Batch automatically handles job scheduling, execution, and resource allocation.
This modular approach is designed to scale. As quantum hardware capacity expands, the workflow can readily distribute these tasks across multiple quantum processing units. so the pipeline remains a robust, scalable solution for large-scale biochemical screening as more powerful quantum resources become available.
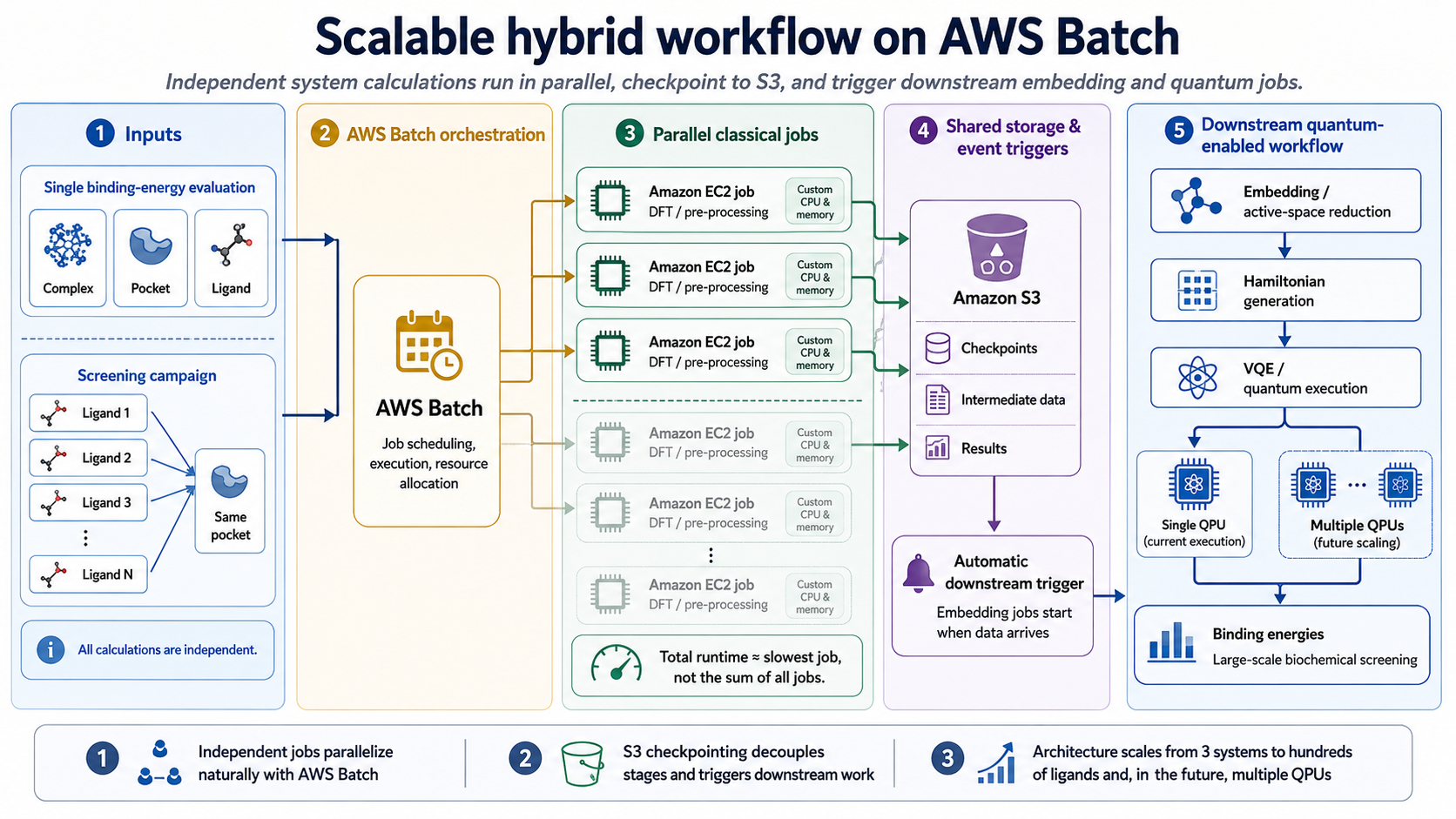
Example scalable AWS Batch architecture for hybrid quantum-classical binding energy calculations. Independent complex, pocket, ligand, and ligand-screening jobs are executed in parallel on Amazon EC2 and checkpointed to Amazon S3. These jobs trigger downstream embedding, Hamiltonian construction, and VQE execution, enabling the workflow to scale from a single binding energy estimate to large screening campaigns.
Conclusion
This work demonstrated that a quantum-classical hybrid pipeline for ligand-protein binding calculations is feasible: binding energies computed through both classical and quantum routes, VQE convergence and reproducibility confirmed across parameter sweeps. The quantum performance surpassed classical DFT calculations for a small molecule example. Since the quantum stage is expressed as a high-level Classiq model, the same workflow can be synthesized for different execution targets, including Braket-accessible hardware, and then retargeted by changing the execution backend as quantum devices become suitable for larger active-space calculations.
The combination of projection-based embedding and scalable cloud HPC means that the hardest part of the computation, characterizing a large, realistic protein system, can be handled classically at whatever scale the problem demands, while the quantum stage remains focused on the fragment most relevant to the binding interaction. This allocation saves computational time and resources by focusing the high-accuracy quantum treatment on the chemically active fragment, while treating the protein environment classically, and without discarding the structural context needed for meaningful binding estimates. As quantum hardware matures and active spaces grow, the classical infrastructure scales with it.
The binding energy pipeline described in this post is part of a growing library of quantum-classical workflows developed on the Classiq platform. Explore related applications:
- Molecule Eigensolver – Uses the Variational Quantum Eigensolver (VQE) to compute ground-state energies of molecules such as H₂, H₂O, and LiH, enabling deeper understanding of chemical properties relevant to drug discovery and materials science.
- Protein Folding – Applies quantum optimization (QAOA) to determine the three-dimensional structure of amino acid chains, a problem of critical importance in medicine and biochemical research.
- Photosynthetic Energy Transfer (FMO Complex) – Simulates quantum interference in energy transfer within the Fenna-Matthews-Olson photosynthetic complex using continuous-time quantum walks, illustrating quantum computing’s potential in modeling biological processes.
For more information on the Classiq platform and how it is used in biochemical processes development or use cases in other industries visit their solution in the AWS Solutions Library or gain access to the Classiq platform via AWS Marketplace. Start experimenting with quantum computing workloads using Amazon Braket or the Classiq platform to get hands on experience with quantum computers.