AWS HPC Blog
Reducing costs by 50% while processing population-scale genomics with Mountpoint for Amazon S3 and AWS Batch
This post was contributed by Kambiz Shahim, Ankit Kalyani, and Chris Wright.
Oxford Nanopore Technologies used Mountpoint for Amazon S3, AWS Batch, and Nextflow to build EPI2ME Cloud, a managed compute environment for processing human genomes at population-scale reliably and securely while reducing computational costs by 50%. EPI2ME Cloud forms Oxford Nanopore’s suite of local and cloud-based analysis platforms, bioinformatics tools, workflows, and resources designed to simplify genomic data processing and interpretation.
In recent years, the promise of precision medicine became a tangible reality. Genomic data, once the domain of small research labs, is now at the heart of national healthcare strategies. Singapore’s National Precision Medicine (NPM) program is an example of this transformation – a comprehensive, long-term initiative to integrate genomic insights into everyday clinical practice.
Oxford Nanopore offers innovative DNA and RNA sequencing platforms that are used in cutting-edge science applications globally, such as outbreak monitoring, cancer research, and analyzing population genetics. Their role in the NPM program focuses on sequencing over 10,000 genomes representing Singapore’s diverse population, including Malay, Indian, and Chinese communities participating in the PRECISE-SG100K population cohort. As an outcome of this human whole-genome sequencing (WGS) exercise, Oxford Nanopore delivers detailed, comprehensive genomic data to advance research and support precision healthcare. This post describes how Oxford Nanopore built and scaled EPI2ME Cloud – all centered on the wf-human-variation, a Nextflow pipeline designed for identifying genomic variations.
Outlining the pipeline
Figure 1 shows the EPI2ME Cloud architecture and pipeline workflow.
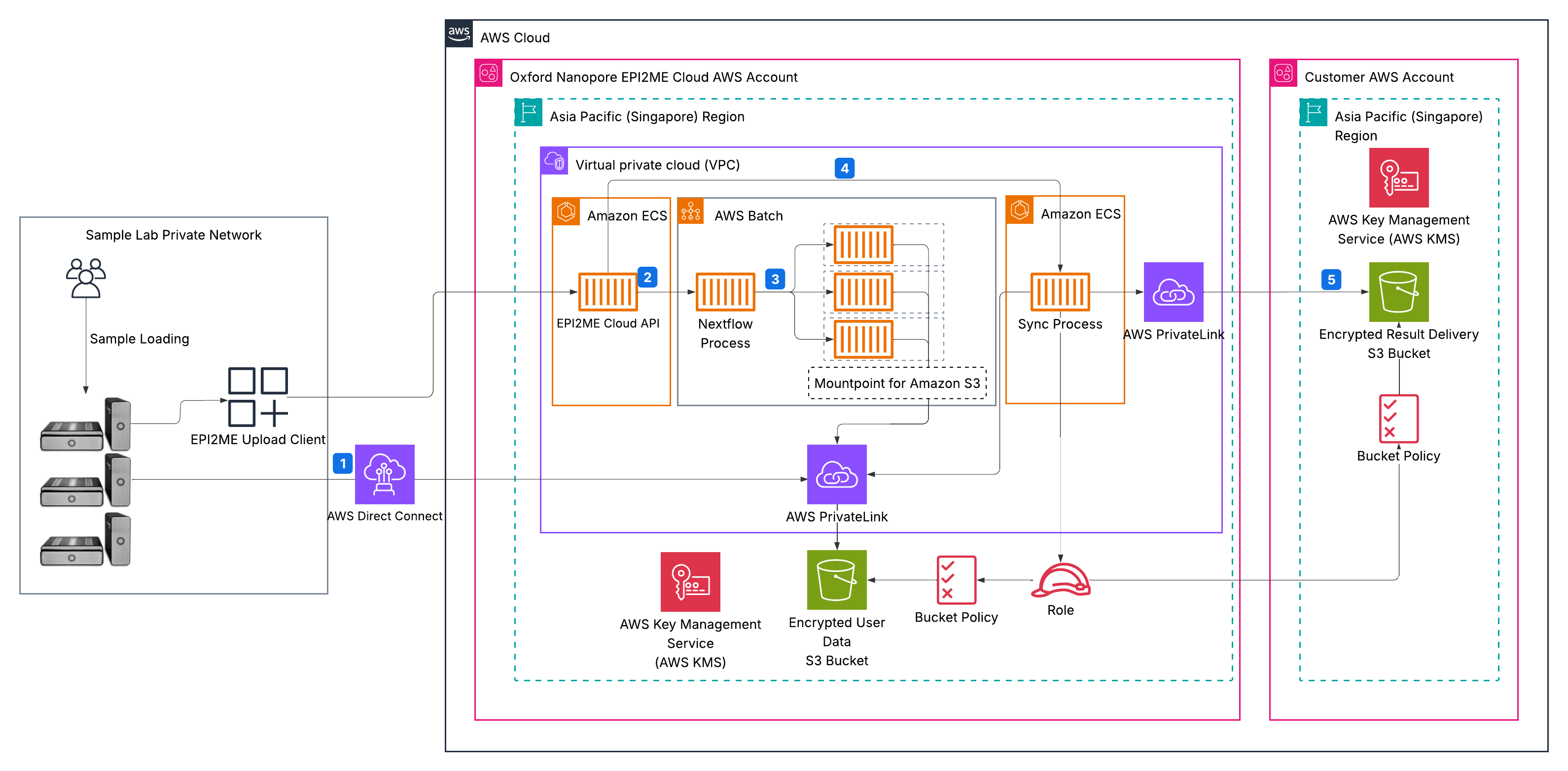
Figure 1: Architecture and workflow of the automated genomics analysis pipeline
The pipeline workflow consists of the following steps:
- Sequencers transfer output data as BAM files directly to an Amazon Simple Storage Service (Amazon S3) bucket using pre-signed upload URLs that EPI2ME Cloud generates. This approach eliminates processing delays and reduces risks from local hardware failures.
- Data arrives in Amazon S3 that starts the orchestration phase. Oxford Nanopore’s EPI2ME Cloud automatically triggers Nextflow pipeline execution by submitting a job to a dedicated AWS Batch queue, configured with the appropriate input paths, genomic references, and workflow parameters.
- Nextflow executes the wf-human-variation workflow using AWS Batch and cost-optimized Amazon Elastic Compute Cloud (Amazon EC2) Spot instances for compute resources.
- Amazon S3 stores the validated results in the customer’s AWS account through a secure synchronization process, ensuring data ownership and security compliance.
Using the wf-human-variation pipeline for variant calling at scale
Built using Nextflow DSL2, the wf-human-variation pipeline is a modular, portable, and reproducible workflow designed to extract the full power of long-read human genomics. At its core, the workflow performs variant calling, the process of identifying genetic and epigenetic differences (variants) between a sample’s DNA sequence and a reference genome. It begins with BAM files (sequencer output) and ends with fully phased and annotated variant calls. In this deployment, the workflow runs entirely on AWS and is scaled horizontally for high-throughput performance by running multiple compute instances in parallel.
For wf-human-variation, Oxford Nanopore rearchitected genome analysis tools, such as Clair3 (a variant calling tool), for horizontal scaling across many small, cost-effective Amazon EC2 Spot instances. This modular pipeline encapsulates each step in its own container using Nextflow DSL2 modules. You can then update or substitute individual steps without disrupting the entire system. Nextflow profiles provide configuration options that you can customize for specific environments.
Scaling on demand by using AWS Batch
Genomics analysis at scale requires elastic compute resources that automatically scale up or down based on workload demand. For Singapore’s NPM program, Oxford Nanopore accomplished this through AWS Batch, a fully managed batch computing service that plans, schedules, and runs containerized batch machine learning (ML), simulation, and analytics workloads.
Each pipeline step runs as a separate AWS Batch job. AWS Batch handles resource provisioning and job placement, while Nextflow orchestrates job submission, monitors execution, and retries failures, creating a pipeline optimized for performance and cost.
In practice, this means that the system can simultaneously process hundreds of genomes, each broken into smaller chunks, and run in parallel. One genome might involve a hundred separate jobs.
Accessing Amazon S3 as a local file system using Mountpoint for Amazon S3
Traditionally, processing genomic data at this scale required staging data into and out of compute instances through either copying data from object storage to instance storage or by using network attached storage with file systems, such as Lustre. For large datasets, this adds time, complexity, and cost. Using Mountpoint for Amazon S3 removed the staging bottleneck in this deployment.
Mountpoint for Amazon S3 is a high-throughput file client for mounting an Amazon S3 bucket as a local file system. Running processes, in this case genomic tools running in AWS Batch jobs, access Amazon S3 objects directly through Mountpoint without downloading them to the instance’s storage.
Oxford Nanopore’s wf-human-variation workflow requires high-throughput and efficient data access. Each genome analysis run comprises approximately 1,400 tasks, with each task requiring 500MB from a 200GB file. Without Mountpoint, processing these genomes would require downloading 77 PB daily from S3 to local instance storage. By eliminating staging requirements, Mountpoint enables each task to pull only the required 500 MB, significantly reducing both local storage costs and CPU idle time.
For developers, this approach is seamless. The pipeline simply mounts the relevant Amazon S3 prefix and executes genomic tools as it would on any local file system. For operation teams, the benefits are substantial: reduced storage costs, simpler workflows, and fewer moving parts.
Delivering results
After the analysis completes, the system collates the results: quality control metrics, phased and annotated gVCF files, and structural variant summaries. These are validated using a simple set of runtime checks and then moved into a designated Amazon S3 bucket in the customer’s AWS account (Precision Health Research, Singapore in this case).
In national precision medicine programs worldwide, patient confidentiality and data sovereignty are paramount. Tightly scoped AWS Identity and Access Management (IAM) roles and pre-established trust relationships ensure this cross-account delivery mechanism is handled securely. Only the data that meets the customer’s approved quality metrics is transferred.
The pipeline captures audit trails throughout with AWS CloudTrail and Amazon S3 server access logging providing full traceability. The system records every pipeline run, ensuring full reproducibility down to the individual genome.
Designing for security and compliance
Data is encrypted at rest and in transit with strict access controls defined in IAM). Mountpoint for Amazon S3 accesses the project’s S3 bucket through AWS PrivateLink for Amazon S3, ensuring data remains within the project’s dedicated Amazon Virtual Private Cloud (Amazon VPC).
The pipeline maintains immutable logs and retains them according to local policy. Sensitive results are not stored in intermediate locations. The pipeline is auditable and reproducible, aligned with legal standards and frameworks.
Showing clear impact with real-world results
In production, this architecture proves highly effective. Throughput scales to hundreds of genomes per week, and the infrastructure can burst higher when required. Median runtimes remain low through intelligent parallelization and the elimination of data staging by Mountpoint for Amazon S3 to eliminate both redundant storage and CPU idle time waiting for data to be staged.
Additionally, the system operates with minimal manual oversight. Sequencing analysis, quality control, and result delivery are all handled automatically, freeing engineers and bioinformaticians for higher-level tasks and ensuring consistency across thousands of samples.
This architecture combines Amazon EC2 Spot instances, Mountpoint for Amazon S3, and AWS Batch to deliver strong cost efficiency. When compared to traditional genomics pipelines with local storage staging, this approach reduces total cost of ownership by 50% while maintaining performance.
Conclusion
By combining Oxford Nanopore’s high-throughput sequencing, EPI2ME’s orchestrated workflows, and AWS’s scalable, secure, and reliable offerings, such as AWS Batch and Mountpoint for Amazon S3, Singapore’s NPM program now runs end-to-end automated Nanopore sequencing for genome analysis at population scale. This collaboration delivers reproducible, secure, and cost-efficient insights, turning raw data into precision medicine outcomes quickly and reliably. To learn more about implementing similar solutions, see the AWS Batch Getting Started Guide and Mountpoint for Amazon S3 documentation.