亚马逊AWS官方博客
在 Amazon SageMaker 上使用 ESMFold 语言模型加速蛋白质结构预测
先决条件
我们建议在 Amazon SageMaker 工作室笔记本电脑中运行这个示例,该笔记本电脑在 ml.r5.xlarge 实例类型上运行 PyTorch 1.13 Python 3.9 CPU 优化的映像。
以视觉形式呈现曲妥珠单抗的实验结构
首先,我们使用 biopython 库和辅助脚本从 RCSB 蛋白质数据库下载曲妥珠单抗结构:
接下来,我们使用 py3Dmol 库将结构可视化为交互式 3D 视觉内容:
下图显示了蛋白质数据库(PDB)中 1N8Z 的 3D 蛋白质结构。在此图中,曲妥珠单抗轻链显示为橙色,重链显示为蓝色(可变区为浅蓝色),HER2 抗原显示为绿色。
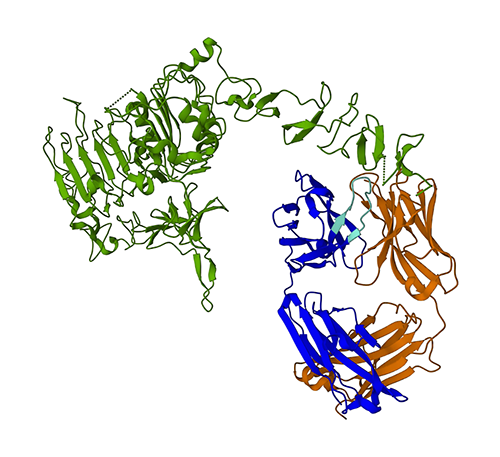
我们将首先使用 ESMFold,根据氨基酸序列预测重链(链 B)的结构。然后,将预测结果与上面显示的实验测定的结构进行对比。
使用 ESMFold 根据曲妥珠单抗重链序列预测其结构
让我们使用 ESMFold 模型来预测重链的结构,并将其与实验结果进行对比。首先,使用 Studio 中预置的 notebook 环境,该环境预装了几个重要的库,如 PyTorch。虽然我们能够使用加速实例类型来提高 notebook 分析的性能,但现在我们改用非加速实例,并在 CPU 上运行 ESMFold 预测。
首先,从 Hugging Face Hub 加载预训练的 ESMFold 模型和标记器:
接下来,将模型复制到我们的设备(本例中为 CPU),并设置一些模型参数:
为了准备用于分析的蛋白质序列,我们需要对其进行标记化处理。这样可以将氨基酸符号(EVQLV…)转换为 ESMFold 模型可以理解的数字格式(6,19,5,10,19,…):
接下来,我们将标记化输入复制到模式中,进行预测,并将结果保存到文件中:
对于非加速实例类型(如 r5),这大约需要 3 分钟。
我们可以通过对比实验结构,来检验 ESMFold 预测的准确性。我们使用密歇根大学 Zhang Lab 开发的 US-Align 工具来完成这项工作:
| PDBchain1 | PDBchain2 | TM-Score |
| data/prediction.pdb:A | data/experimental.pdb:B | 0.802 |
模板建模得分(TM-score)是评测蛋白质结构相似性的指标。得分为 1.0 表示完全匹配。得分高于 0.7 表示两个蛋白质具有相同的骨干结构。得分高于 0.9 表示蛋白质在下游使用中具有功能互换性。在我们的示例中,TM-Score 达到 0.802,表示 ESMFold 预测可能适用于结构评分或配体结合实验等应用,但可能不适用于分子置换等需要极高精度的使用案例。
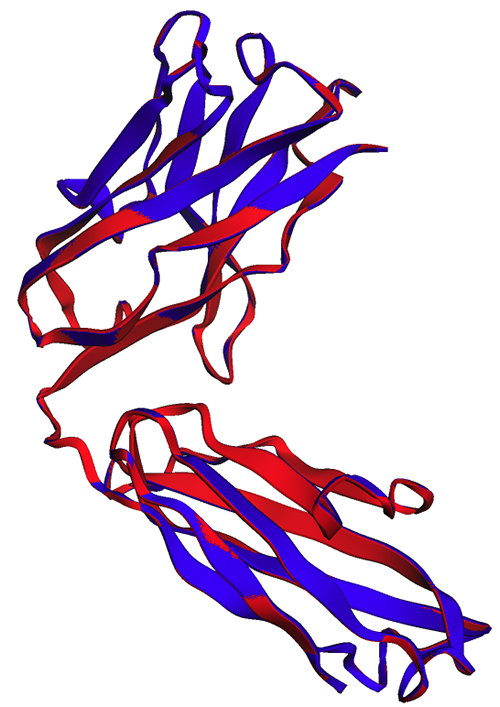
我们可以通过可视化对齐结构来验证这一结果。这两个结构的重叠程度很高,但并不完全重叠。蛋白质结构预测是一个快速发展的领域,许多研究团队都在开发越来越精确的算法!
将 ESMFold 部署为 SageMaker 推理端点
在 notebook 中运行模型推理可用于实验目的,但如果需要将模型与应用程序集成呢? 或者是与 MLOps 管道集成呢? 在这种情况下,更好的选择是将模型部署为推理端点。在下面的示例中,我们将 ESMFold 作为 SageMaker 实时推理端点部署在加速实例上。SageMaker 实时端点提供了一种可扩展、经济高效且安全的方式来部署和托管机器学习(ML)模型。通过自动扩缩,您可以调整运行端点的实例数量以满足应用程序的需求,从而优化成本并确保高可用性。
利用预置的 Hugging Face 的 SageMaker 容器,可以轻松地为常见任务部署深度学习模型。然而,对于像蛋白质结构预测这样的新使用案例,我们需要定义一个自定义的 inference.py 脚本,来加载模型、运行预测和格式化输出内容。该脚本包含的代码与我们在 notebook 中使用的代码基本相同。我们还创建了一个 requirements.txt 文件来定义一些 Python 依赖关系,供我们的端点使用。您可以在 GitHub 存储库中看到我们创建的文件。
在下图中,曲妥珠单抗重链的实验(蓝色)和预测(红色)结构非常相似,但并不完全相同。

在 code 目录中创建了必要的文件后,我们使用 SageMaker HuggingFaceModel 类部署模型。该类使用一个预置的容器,简化将 Hugging Face 模型部署到 SageMaker 的过程。请注意,创建端点可能需要 10 分钟或更长时间,具体取决于我们区域中 ml.g4dn 实例类型的可用性。
完成端点部署后,我们可以重新提交蛋白质序列,并显示预测的前几行内容:
由于我们将端点部署到加速实例中,因此预测应该只需几秒钟。结果中的每一行对应于一个原子,包括氨基酸标识、三个空间坐标和表示该位置预测置信度的 pLDDT 分数。
| PDB_GROUP | ID | ATOM_LABEL | RES_ID | CHAIN_ID | SEQ_ID | CARTN_X | CARTN_Y | CARTN_Z | OCCUPANCY | PLDDT | ATOM_ID |
| ATOM | 1 | N | GLU | A | 1 | 14.578 | -19.953 | 1.47 | 1 | 0.83 | N |
| ATOM | 2 | CA | GLU | A | 1 | 13.166 | -19.595 | 1.577 | 1 | 0.84 | C |
| ATOM | 3 | CA | GLU | A | 1 | 12.737 | -18.693 | 0.423 | 1 | 0.86 | C |
| ATOM | 4 | CB | GLU | A | 1 | 12.886 | -18.906 | 2.915 | 1 | 0.8 | C |
| ATOM | 5 | O | GLU | A | 1 | 13.417 | -17.715 | 0.106 | 1 | 0.83 | O |
| ATOM | 6 | cg | GLU | A | 1 | 11.407 | -18.694 | 3.2 | 1 | 0.71 | C |
| ATOM | 7 | cd | GLU | A | 1 | 11.141 | -18.042 | 4.548 | 1 | 0.68 | C |
| ATOM | 8 | OE1 | GLU | A | 1 | 12.108 | -17.805 | 5.307 | 1 | 0.68 | O |
| ATOM | 9 | OE2 | GLU | A | 1 | 9.958 | -17.767 | 4.847 | 1 | 0.61 | O |
| ATOM | 10 | N | VAL | A | 2 | 11.678 | -19.063 | -0.258 | 1 | 0.87 | N |
| ATOM | 11 | CA | VAL | A | 2 | 11.207 | -18.309 | -1.415 | 1 | 0.87 | C |
使用与以前相同的方法,我们可以看到 notebook 和端点的预测结果完全相同。
| PDBchain1 | PDBchain2 | TM-Score |
| data/endpoint_prediction.pdb:A | data/prediction.pdb:A | 1.0 |
如下图所示,在 notebook 中生成的 ESMFold 预测结果(红色)与端点生成的预测结果(蓝色)完全一致。
清理
为了避免产生进一步的费用,我们删除了推理端点和测试数据:
小结
蛋白质结构计算预测是了解蛋白质功能的重要工具。除基础研究外,AlphaFold 和 ESMFold 等算法在医学和生物技术领域也有很多应用。利用这些模型生成的结构洞察,有助于我们更好地了解生物分子是如何相互作用的。这样就能为患者提供更好的诊断工具和疗法。
在这篇文章中,我们将展示如何使用 SageMaker 将 Hugging Face Hub 的 ESMFold 蛋白质语言模型部署为可扩展的推理端点。有关在 SageMaker 上部署 Hugging Face 模型的更多信息,请参阅结合使用 Hugging Face 与 Amazon SageMaker。您还可以在 Awesome Protein Analysis on AWS GitHub 存储库中找到更多蛋白质科学示例。如果您还想了解其他示例,请给我们留言!
关于作者
 Brian Loyal 是 Amazon Web Services 全球医疗保健和生命科学团队的高级人工智能/机器学习解决方案架构师。他在生物技术和机器学习领域拥有超过 17 年的经验,热衷于协助客户解决基因组和蛋白质组学方面的难题。在业余时间,他喜欢烹饪,享受与朋友和家人的就餐时光。
Brian Loyal 是 Amazon Web Services 全球医疗保健和生命科学团队的高级人工智能/机器学习解决方案架构师。他在生物技术和机器学习领域拥有超过 17 年的经验,热衷于协助客户解决基因组和蛋白质组学方面的难题。在业余时间,他喜欢烹饪,享受与朋友和家人的就餐时光。
 Shamika Ariyawansa 是 Amazon Web Services 全球医疗保健和生命科学团队的人工智能/机器学习专业解决方案架构师。他热衷于与客户合作,通过提供技术指导,促进客户在 AWS 上进行创新和构建安全的云解决方案,从而加快客户采用人工智能和机器学习的速度。工作之余,他喜欢滑雪和越野运动。
Shamika Ariyawansa 是 Amazon Web Services 全球医疗保健和生命科学团队的人工智能/机器学习专业解决方案架构师。他热衷于与客户合作,通过提供技术指导,促进客户在 AWS 上进行创新和构建安全的云解决方案,从而加快客户采用人工智能和机器学习的速度。工作之余,他喜欢滑雪和越野运动。
 Yanjun Qi 是 AWS Machine Learning 解决方案实验室的高级应用科学经理。她通过创新和运用机器学习,协助 AWS 客户加快采用人工智能和云技术的速度。
Yanjun Qi 是 AWS Machine Learning 解决方案实验室的高级应用科学经理。她通过创新和运用机器学习,协助 AWS 客户加快采用人工智能和云技术的速度。