亚马逊AWS官方博客
基于亚马逊云科技 CI/CD 和 Amazon Batch 服务构建云端单细胞分析流程
单细胞分析流程概述
长期以来人们针对人体机能的认知和疾病病因的研究都是基于组织或者血液的这样一种整体概念。目前人们所熟悉的二代测序结果是在大量细胞的平均水平上对整个细胞群体进行组学分析,但是这样仅能反映细胞群体中的平均化信号和主流信号。细胞作为生物体结构和功能的基本单位,平均化的结果忽略了单个细胞间的个体差异信息,对于神经生物学、自身免疫疾病、发育研究、肿瘤研究等涉及到细胞间异质性的研究来说,是远远不够的。单细胞测序技术作为现代应用和临床医学的整合,能够在单个细胞的水平上通过检测SNV、CNV、单细胞基因组结构变异、基因表达水平、基因融合、单细胞转录组的选择性剪接和单细胞表观基因组的DNA甲基化来识别出特定细胞的类型和功能, 健康或状态变化变异,进而实现在单细胞层面上对疾病和生物学过程中的遗传特征进行研究。
由于本地计算集群的资源在弹性和扩展性上有一定的限制,在大批量样本的生产场景下, 集群运维的复杂性和不可控的任务排队等候会极大影响运营项目交付效率. 本文以10X Cell Ranger单细胞测序分析流程软件为例, 旨在以介绍通过Cromwell的网页提交分析任务,调度Amazon Batch服务提供计算资源, 并辅以亚马逊云科技的CI/CD服务构建更新分析容器镜像。
本文涉及主要软件和服务介绍
Cromwell介绍
Cromwell 是由Broad Institute 开发的Workflow系统,通过 Cromwell 工作流管理系统用户可以将 WDL(Workflow Define Language)描述的workflow任务转化为Amazon Batch的任务,每个任务交由后台的Amazon ECS编排调度的容器来运行,用户只需为后台实际调用的计算和存储等服务付费,充分利用亚马逊云科技带来的弹性和高扩展性。在计费方便计算实例也同时支持按需,预留实例和SPOT方式来帮用户降低成本。
Cell Ranager
Cell Ranger是10X genomics公司专门针对单细胞转录组测序分析开发的数据分析软件,将fastq测序数据比对到参考基因组,输出表达定量矩阵,再通过降维(pca),聚类(Graph-based& K-Means)得到可视化(t-SNE)结果,结合配套的Loupe Cell Browser给予研究者更多探索单细胞数据的机会。Cell Ranger的高度集成化,使得单细胞测序数据探索变得更加简单,研究者有更多的时间来做生物学意义的挖掘。
Amazon Batch概述
Amazon Batch服务作为可用于任意规模完全托管的批处理服务,在生命科学行业Amazon Batch 让开发人员、生信专家和工程师能够轻松高效地在亚马逊云科技平台上运行成千上万个批处理计算作业。Amazon Batch 可根据提交的批处理作业的卷和特定资源需求动态预置最佳的计算资源(如 CPU 或内存优化实例)数量和类型。借助 Amazon Batch,您无需安装和管理运行您的作业所使用的批处理计算软件或服务器集群,从而使您能够专注于分析结果和解决问题。
流程架构图

测试环境准备
准备网络环境
网络环境我们可以通过提前创建好的CloudFormation模版aws-vpc.template.yaml来创建,用户也可以通过VPC创建向导进行VPC和子网的创建,在创建过程中根据实际需求创建VPC CIDR、公有子网、私有子网和路由等信息。
准备Amazon EFS文件系统
在本篇文章中我们将创建EFS文件系统用于后续用户存放单细胞分析中所需要的参考基因组。注意修改Amazon EFS关联安全组允许VPC内部的实例连接EFS文件系统。

准备Amazon Batch环境
这里我们将通过cn-gwfcore-root.templete模板来创建 Batch环境,这里包括计算资源使用的启动模版、所属网络、计算环境和任务队列等信息,CloudFormation输出主要资源如下:

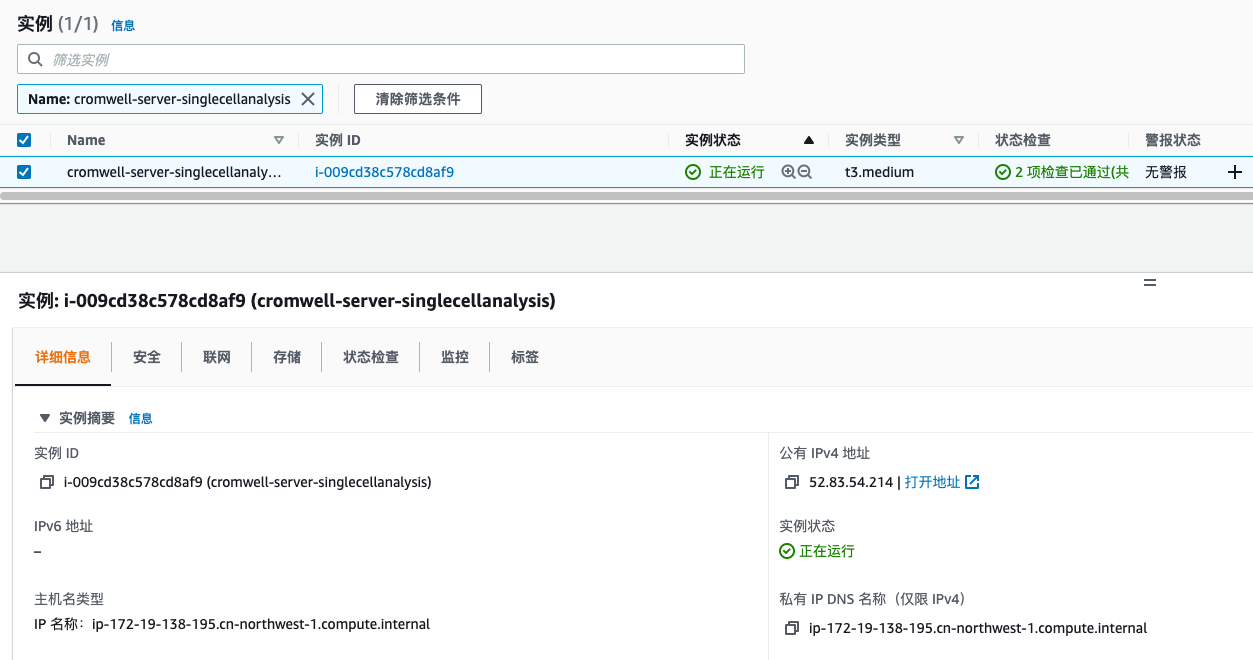
准备Cromwell Server
通过Cromwell-resource CloudFormation模版启动Cromwell Server,在配置页面我们需要配置实例类型、网络配置、安全组和软件版本信息(我们提前将cromwell server 70版本下载到存储桶)等,填写完所需配置后点击下一步完成Cromwell Server的快速部署。
SSH登录Cromwell Server, 我们可以检查cromwell配置文件/home/ec2-user/cromwell.conf. backend配置如下:
通过https访问Cromwell Server公有 IPv4 DNS地址,忽略证书提示后可以看到如下页面, 后续我们会从此页面提交单细胞分析任务的和任务查看。

测试数据准备
(1) 通过10x genomics官网下载对应物种的参考基因组数据(本示例以小鼠参考基因组为例)并上传至Amazon EFS文件系统。这里我们可以通过登录Cromwell Server实例并参考文档挂载EFS目录并将数据拷贝至EFS根目录。
(2) 准备好Cell Ranger安装包到Amazon S3存储桶(用户下载需要Token所以用户可以下载到自己指定的公开路径)。
(3) 上传fastq基因测序数据至Amazon S3指定目录。
CICD流程
创建ECR镜像库
登录亚马逊云科技管理控制台,选择ECR服务,点击创建存储库,输入存储库名称,点击创建存储库完成创建。这里设置镜像仓库名称为singlecellanalysis, 如下图所示
准备Dockerfile
用户根据自己的业务需求构建自己的Dockerfile
创建buildspec.yml
创建buildspec.yml文件用于项目构建,主要任务为登录ECR,构建Docker镜像并推送至ECR。内容如下:
创建CodeCommit项目
(1) 通过登录亚马逊云科技管理控制台,选择CodeCommit服务, 点击创建存储库,在配置页面输入存储库名称和描述信息后完成创建。
(2) 参考文档进行初始化是指克隆存储库。
(3) 将Dockerfile,buildspec.yml和其他所需文件推送至CodeCommit对应的存储库。

创建CodeBuild项目
(1)登录亚马逊云科技管理控制台,选择CodeBuild服务, 点击创建项目。
(2)在项目配置页面输入项目名称,源配置页面源提供商选择Amazon CodeCommit, 并选择对应的存储库和分支。
(3)环境配置页面配置如下

(4)Buildspec 配置选项出构建规范选择使用buildspec文件,其他保持默认即可,点击页面底端创建构建项目完成构建。
(5)这里需要注意的是默认的ServiceRole无法访问ECR镜像,这里我们去需要登录IAM服务,在角色页面输入codebuild找到我们对应的角色,点击添加权限并添加AmazonEC2ContainerRegistryFullAccess策略至该角色。
(6)切换至CodeBuild服务并选择构建项目,点击右上角的开始构建完成项目构建。
(7)观察CodeBuild的构建日志,待项目完成构建我们登录ECR检查确认最新镜像已经构建完毕并推送至ECR镜像库。

提交分析任务
在后续任务提交的时候我们需要提交两个文件:
input.json文件中需要配置样本名称(cellranger_count_GEX.sample_name),期望细胞数(cellranger_count_GEX.expect_cells),fastq文件路径(cellranger_count_GEX.fq1s,cellranger_count_GEX.fq2s),参考基因组路径(cellranger_count_GEX.transcriptome),及对应的Amazon Batch队列和Docker镜像地址。
workflow.wdl中定义:
RuntimeAttr,创建task的runtime结构体(cpu,memory,docker,queue),方便wdl内部定义默认值,同时也接受inputs.json外部输入值
workflow定义流程的输入,输出,runtime和task的调用
task定义流程中的任务具体执行内容,定义任务的输入输出,命令和runtime
参考示例如下
任务提交
在日常的工作中客户生信工程师需要编写WDL格式的基因分析流程脚本,通过网页方式访问Cromwell Server并通过API的方式提交任务。用户也可以使用命令行方式或者三方工具类似Postman的方式提交任务。
Cromwell Server网页方式提交:

命令行方式提交:
curl -X POST "https://ec2-52-83-54-214.cn-northwest-1.compute.amazonaws.com.cn/api/workflows/v1" -H "accept: application/json" -H "Content-Type: multipart/form-data" -F "workflowSource=@workflow.wdl" -F "workflowInputs=@inputs.json;type=application/json"
任务状态查看
用户通过网页API的方式


命令行方式根据任务ID查看任务状态
curl -X GET "https://ec2-52-83-54-214.cn-northwest-1.compute.amazonaws.com.cn/api/workflows/v1/d7fb3382-7c1e-4829-b4f8-1796da81d85f/status" -H "accept: application/json"
用户也可以通过Amazon Batch Console查看任务状态,执行的日志可以也可以通过CloudWatch日志组来查看任务运行

任务输出
用户可以通过API或者命令行的方式来查看任务输出,输出内容如下:
结果输出
分析结果最终会存放到Amazon S3中,这里我们需要关注的输入中的html 报告文件和h5ad表达矩阵文件,在报告文件中我们一般关注如下指标作为样本和实验质量标准:
- 每个细胞基因数的中间数(Median genes per cell)> 1000
- UMI 校正后匹配的UMI数量valid barcode > 90%
- Q30 Bases in barcode/RNA read/UMI: 大于30%
- 与细胞相关的UMI可靠地比对到基因组Fraction reads > 70%
另外一个比较重要的就是UMI-barcodes的曲线图,曲线图上的蓝色区域代表真细胞,灰色区域代表背景噪音,如下图所示。

用户可以以h5ad为数据源,使用开源的分析软件(scanpy/seurat)进行下游分析,例如,降维,聚类,差异基因表达分析等。

总结
用户通过CloudFormation快速完成环境部署,用户可以通过网页/命令行/三方快捷的提交任务进行单细胞流程的分析。后端借助于亚马逊云科技提供的海量弹性资源快速有效的完成单细胞分析用于下游分析。
参考资料
Cromwell on AWS: https://docs.opendata.aws/genomics-workflows/orchestration/cromwell/cromwell-overview.html
Github: https://github.com/qiufengliu/singlecellanalysis