Blog de Amazon Web Services (AWS)
Diagnóstico de leucemia mediante machine learning en el Laboratorio de Leucemia de Múnich con Amazon SageMaker
Por Erika Pelaez Coyotl, Data Scientist en Machine Learning Solutions Lab;
James Golden, PhD Data Scientist en Machine Learning Solutions Lab team;
Tamas Madl, PhD en Mission Solutions Specialist en Amazon Web Services;
Niroshan Nadarajah, lead Bioinformatician and Innovation manager en Munich Leukemia Laboratory y
Liwen You, PhD Data Scientist en Machine Learning Solutions Lab.
- Manejo de archivos de Variant Call Format de NGS para generar funciones útiles para la clasificación
- Exploración de la separabilidad de las diferentes clases en función de métricas de similitud
- Búsqueda del mejor modelo para la clasificación e interpretabilidad de los subtipos de leucemia
- Conclusiones de la colaboración
Introducción
Según el estudio Global Burden of Disease de 2018, la leucemia se refiere a un grupo de cánceres de la sangre que representan un desafío importante para la atención médica y causan más de 310 000 muertes al año en todo el mundo. El diagnóstico y el tratamiento son muy difíciles debido a su heterogeneidad: hay 31 entidades de leucemia (subtipos) definidas por la Organización Mundial de la Salud a partir de 2017.
En la actualidad, la leucemia se diagnostica mediante una combinación de métodos. Estos métodos requieren de equipos complejos y científicos y técnicos de laboratorio clínico altamente calificados, ambos recursos escasos, lo que aumenta los tiempos de respuesta y los costos. El tiempo promedio desde la recepción de la muestra hasta la generación del informe puede tardar hasta diez días.
Un cambio de paradigma en la metodología diagnóstica
La secuenciación de próxima generación (NGS), una tecnología de secuenciación de ADN masiva en paralelo que es capaz de leer un genoma humano completo en un día, ha demostrado ser muy prometedora para identificar subtipos de leucemia, así como para indicar un tratamiento más específico y eficaz.
El Dr. Prof. Torsten Haferlach, CEO de MLL, dijo: “Para establecer este cambio de paradigma y pasar de una revisión principalmente manual de las características morfológicas por parte de expertos con amplia experiencia, a un enfoque objetivo y algorítmico que utiliza características basadas en la genética molecular, MLL se asoció con el Laboratorio de soluciones de Amazon ML para abordar esta extraordinaria hazaña. Este trabajo conducirá a encontrar más curas y prolongará la vida de todos los pacientes”.
Clasificación del cáncer basada en datos de NGS mediante machine learning
El Laboratorio de soluciones de Amazon Machine Learning asumió el reto de ayudar a MLL a desarrollar un modelo innovador de apoyo a las decisiones clínicas basado en un conjunto de datos de secuenciación de próxima generación (NGS). Los datos de NGS son de dimensiones extremadamente grandes y, por lo general, están compuestos por cantidades muy pequeñas de datos de entrenamiento de calidad. MLL, tras haber completado el proyecto de 5000 genomas, tiene uno de los conjuntos de datos más grandes de este dominio. La mayoría de las muestras de los 5000 pacientes con leucemia involucrados se secuenciaron mediante la secuenciación del genoma completo (WGS) y la secuenciación del transcriptoma completo (WTS), y se diagnosticaron mediante los métodos de referencia de la OMS de diagnóstico ortogonal.
Preparación de datos
Con el alcance global de la investigación de MLL, es imperativo que los datos se almacenen en servicios en la nube que sean seguros y cumplan con las normas de privacidad. AWS está disponible en muchos países y regiones, lo que ofrece comodidad y bajo costo para los clientes. Todos los datos de los clientes se pueden almacenar y se puede acceder a ellos en cualquier región de AWS. En este caso particular, los datos de MLL se almacenaron en buckets de Amazon S3 dentro de la región seleccionada por el cliente en Fráncfort para cumplir con los requisitos de los datos de atención médica.
Los datos de NGS de ADN (secuenciación del genoma completo) y ARN (secuenciación del transcriptoma completo) se procesaron inicialmente por MLL mediante una canalización personalizada. De la WGS se extraen distintos tipos de variaciones: variación de nucleótido único (SNV), variación estructural (SV) y variación de número de copias (CNV). Los datos de SV y CNV están en archivos VCF, que tienen una estructura compleja. Los archivos VCF incluyen información de variaciones, como la ubicación del cromosoma y el segmento de la variación, la cobertura de lectura y los criterios de filtrado de variaciones. Los archivos VCF para los datos SV y CNV no se pueden importar fácilmente a un formato que puedan incorporar los modelos de machine learning. Se necesita un procesamiento adicional para extraer y agregar características en función del nivel de detalle elegido, que podría ser a nivel de genes, bandas cromosómicas o cromosomas.
A partir de la canalización de WTS, se pueden obtener datos de expresión génica (GE) y fusión génica (GF). La ventaja de los datos de GE es que ya tienen un formato tabular donde cada fila es un paciente y cada columna es el valor de expresión de un gen, por lo que no hay otras transformaciones y solo se debe llevar a cabo un procedimiento de normalización rápido. La GF es más similar a los archivos obtenidos para WGS, por lo que se realizan transformaciones similares.
Establecimos una canalización que toma los archivos de datos de cada paciente y los transforma en un vector de características de cada modalidad. Para los archivos SV, se extrajeron cinco tipos de variaciones: inserciones, eliminaciones, duplicaciones, translocaciones e inversiones a nivel genético para segmentos cortos. Para segmentos más largos, se extrajeron características según el nivel de la banda cromosómica o el cromosoma. Este enfoque también se utilizó para los archivos CNV. Para los datos de WTS, la expresión génica se extrae en forma de recuentos de lecturas y se normaliza siguiendo el método de normalización de los valores M de la media recortada ponderada (TMM). Como resultado, generamos más de 70 000 características a partir de las cinco tablas originales (SNV, SV, CNV, expresión génica, fusión génica). Los datos que faltaban se imputaban de forma diferente según el tipo de fuente. Para algunos tipos, utilizamos el valor más bajo (CNV) o cero (SNV, SV, expresión génica). Al final del procesamiento de datos, cada paciente posee la misma cantidad de características, que se pueden usar como entrada para un modelo de machine learning.
Evaluamos varias estrategias para combinar estas diferentes modalidades, incluido el apilamiento o ensamblaje de modelos entrenados en cada modalidad individual. Sin embargo, el manejo por separado de estos tipos de datos no tiene en cuenta la observación de que existen interacciones no lineales entre características de diferentes modalidades (por ejemplo, conexiones entre la amplificación/eliminación del número de copias y la regulación del aumento o la disminución del nivel de expresión génica, SV asociados con expresión génica aberrante). El modelo tiene que ser capaz de identificar que tales interacciones funcionen bien, por lo que la concatenación de todos los tipos de características para permitir que el modelo aprenda las interacciones entre ellas condujo a una mayor precisión predictiva.

Figura 1: Descripción general de los archivos que se procesaron para obtener un conjunto de datos tabular para el entrenamiento. “||” se refiere a la concatenación de los vectores obtenidos para cada tipo de archivo.
Análisis previo al modelo
El conjunto de datos de MLL contiene pacientes con 30 subtipos diferentes de leucemia. Algunos de los subtipos son mucho más frecuentes que otros, lo que da como resultado un conjunto de datos muy desequilibrado.
 Figura 2: Número de pacientes por subtipo de leucemia.
Figura 2: Número de pacientes por subtipo de leucemia.
Con todos los diferentes tipos de datos convertidos a formato tabular, iniciamos un proceso de ingeniería de características que implicaba la colaboración con expertos de MLL. Revisaron la literatura para encontrar biomarcadores importantes asociados con cada subtipo de leucemia. Con esto en mente, diseñamos un proceso para agregar y combinar las características originales en biomarcadores.
El conjunto de datos final tenía aproximadamente 4500 filas que representaban a los pacientes y 800 columnas que contenían los datos de biomarcadores extraídos, combinados con otras características importantes para cada tipo de datos (CNV, SNV, SV, etc.) fuera del conjunto original de características de los biomarcadores. En otras palabras, el genoma y el transcriptoma de cada paciente estaban representados por un vector con 800 entradas.
Entre los 30 subtipos de leucemia, algunos de ellos están más cerca unos de otros que del resto. Por ejemplo, la MPAL, la AML y la BCP-ALL son muy similares; a veces, el perfil genético de un paciente con MPAL puede ser más similar al de un paciente con BCP-ALL que al de otro paciente con MPAL. Esto presenta un desafío tanto para el algoritmo de machine learning como para los expertos humanos a la hora de diferenciarlos. Para investigar qué tan similares eran los subtipos de leucemia entre sí, calculamos las distancias entre cada paciente y el resto de las muestras y registramos cuáles eran los dos vecinos más cercanos. Luego, agregamos los vecinos por subtipo de leucemia y descubrimos que, de hecho, en algunas de las clases, menos del 20 % de los pacientes en ese subtipo eran muy similares entre sí. Tal como se muestra en la Figura 3. Si encontramos el círculo MPAL, podemos ver que solo el 13 % de todos los pacientes con MPAL son similares entre sí, y que la mayoría de los pacientes similares pertenecen a la AML y la BCP-ALL.
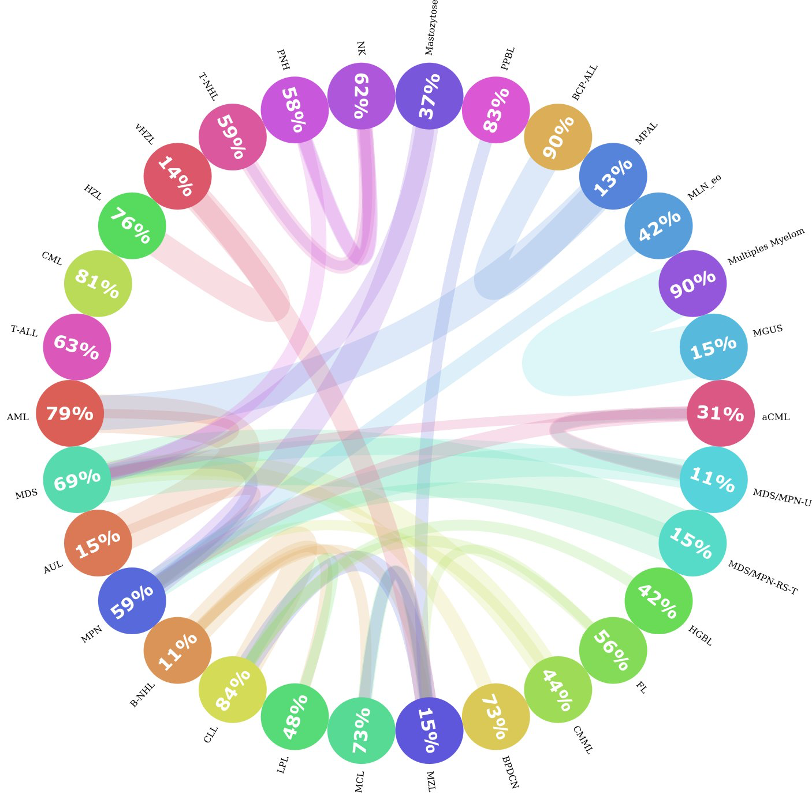
Figura 3: Esquemas circulares de similitudes de las entidades. Esta figura muestra la similitud entre sí de los subtipos de leucemia. Cada círculo representa un subtipo de leucemia y el número dentro del círculo ilustra el porcentaje de vecinos que son del mismo subtipo. Si hay una parte significativa (>= 10 %) de pacientes que son más parecidos a los pacientes de otra entidad, se dibuja una conexión (línea de color) de una entidad a otra. El grosor de la línea representa el tamaño de la parte de pacientes que era similar a otros grupos. El color de la línea representa la dirección de la conexión, es decir, los pacientes de una entidad que pertenecen a otros grupos; se representa con el color de la entidad original. Por ejemplo, los pacientes de la clase MGUS eran más similares a los pacientes con mieloma múltiple. Pero no hay conexiones significativas de forma inversa.
Modelado
Para predecir los subtipos de leucemia de los pacientes, entrenamos un clasificador de varias clases. Con el clasificador y la canalización de extracción de características, creamos un prototipo de sistema que determina automáticamente el subtipo de leucemia del paciente en función de los datos de WGS y WTS mediante Blocs de notas de Amazon SageMaker. Amazon SageMaker es una plataforma de machine learning en la nube que se puede utilizar para crear, entrenar e implementar modelos para prácticamente cualquier caso de uso. Las instancias de bloc de notas proporcionan entornos flexibles para crear modelos de machine learning, lo que ahorra tiempo y esfuerzo a los usuarios a la hora de administrar la infraestructura informática subyacente.
Utilizamos LightGBM, un algoritmo de potenciación de gradiente, para crear nuestro primer clasificador. También utilizamos trabajos de optimización de hiperparámetros (HPO) de SageMaker para ajustar los hiperparámetros de nuestro modelo, un enfoque que encuentra automáticamente la configuración óptima de cualquier algoritmo sin esfuerzo manual mediante el uso de la optimización Bayesiana. Con la HPO, el rendimiento de nuestro modelo LGBM alcanzó una precisión del 82 % en la validación cruzada de 5 iteraciones. Para algunas entidades con biomarcadores únicos, la precisión es muy alta. La CML tiene un gen de fusión BCR-ABL casi único como biomarcador y el modelo LGBM alcanza una precisión del 97 % para la CML.
Sin embargo, para entidades con muy pocos pacientes, con una fuerte similitud con otras entidades o con aquellas que requieren un análisis de laboratorio adicional durante el diagnóstico tradicional, el modelo no funciona tan bien. Para tratar muestras pequeñas y desequilibradas, puede utilizar la generación de datos sintéticos. Probamos diferentes métodos, incluidos LoRAS y ADASYN, pero no vimos ninguna mejora en el modelo.
Los modelos que utilizan el aprendizaje de pocos intentos también se han utilizado para problemas de clasificación con muy pocas muestras para una categoría. Implementamos una arquitectura de aprendizaje de pocos intentos para la predicción del tipo de enfermedad, pero no vimos ninguna mejora. Es probable que esto se deba al desafío de conocer la distribución de un tipo de leucemia con el número muy pequeño de pacientes que teníamos disponibles para cada tipo.
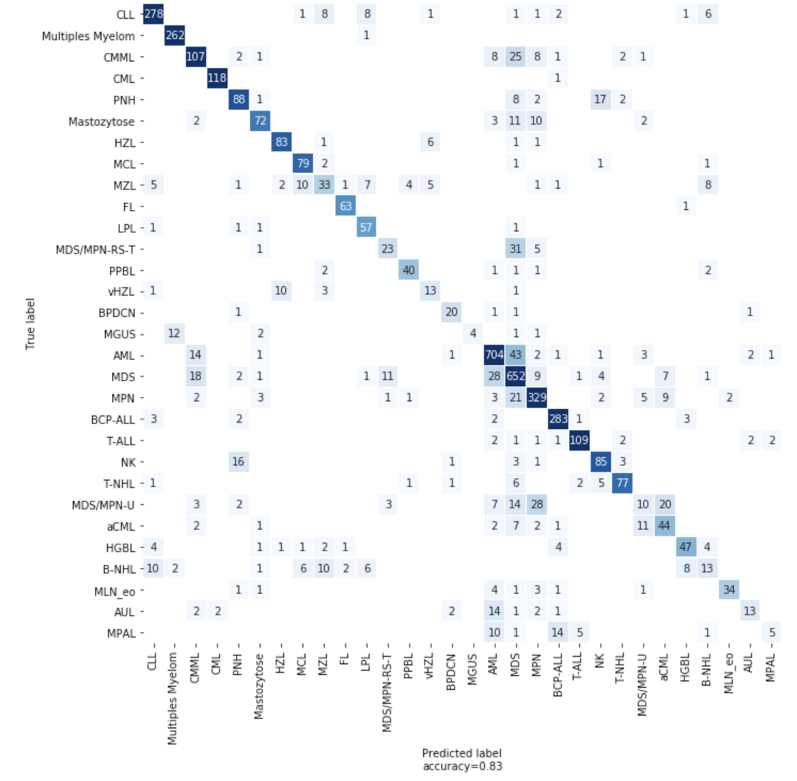
Figura 4: Matriz de confusión que muestra el número de predicciones correctas e incorrectas del modelo
Explicación
Cuando se utiliza un modelo de machine learning para hacer una predicción, comprender por qué se realizó la predicción es tan importante para los investigadores de MLL como la predicción en sí. Para entender por qué el modelo hace predicciones particulares, aprovechamos la biblioteca SHAP de Python. Con esta biblioteca, podemos analizar una predicción del modelo entrenado y obtener información sobre las características utilizadas para realizar la clasificación. Aplicamos SHAP a nuestro modelo LGBM y analizamos los impactos de las características tanto a nivel de la muestra como a nivel global. La Figura 5 ilustra una aplicación de SHAP a nivel de paciente para dos personas diagnosticadas con CML en comparación con la cohorte de CML predicha correctamente. Al utilizar una gráfica de decisión, podemos observar qué características son las que más contribuyen a la predicción del modelo.

Figura 5:gráfico de fuerza de SHAP para un paciente con CML pronosticada correctamente, y un gráfico de decisión para cada paciente con CML, 117 pronosticados correctamente (líneas amarillas) y uno pronosticado incorrectamente (línea roja). Las características se muestran en el eje y en orden de importancia media para todos los pacientes con CML; observe las características conocidas de BCR/ABL1 en la parte superior. Estas características orientaron al clasificador hacia la CML para ambos pacientes, no obstante, para el paciente que se predijo incorrectamente, las características de expresión génica hicieron que el clasificador predijera que el paciente tenía BCP-ALL. Estas gráficas de decisión son útiles para entender cuánto contribuye cada entidad a la clasificación. Tenga en cuenta que el eje x es “probabilidades logarítmicas”, por lo que una entidad que orienta la salida de 0 a 10 es mucho más importante que una entidad que orienta la salida de -5 a 0.
Conclusiones y perspectivas
Colaboración con expertos en el campo: esto ha sido crucial y seguirá siendo una parte clave de la aplicación de machine learning e IA en el ámbito médico. Además de compartir el conjunto de datos más grande y de mayor calidad de su tipo, MLL facilitó la creación del modelo de machine learning al aportar su amplia experiencia en el campo en cada paso del camino. Al principio del proyecto, codificamos nuestro espacio de características a nivel genético y solo logramos una precisión del 67 % en 30 predicciones de subtipos de leucemia. Mediante el uso de los biomarcadores y las características genéticas seleccionadas manualmente por MLL, que incorporan al modelo lo que ya conocen los expertos humanos sobre este campo, logramos una precisión del 77 %. Más tarde, agregamos más características al espacio de características y aumentamos aún más el rendimiento del modelo en un 5 %.
Utilizar las herramientas adecuadas: trabajar con SageMaker aceleró significativamente este proyecto. Dado SageMaker es una plataforma completamente administrada e integrada para machine learning, se permitió que el equipo se centrara en el trabajo de ciencia de datos sin tener que preocuparse por la administración de la infraestructura. La característica integrada de ajuste automático del modelo aumentó la precisión del modelo sin tener que ajustar manualmente la arquitectura del modelo o los hiperparámetros, lo que ahorró tiempo y optimizó el modelo.
Interpretabilidad y capacidad de explicación: MLL continúa innovando y mejorando su enfoque y su visión hacia un diagnóstico de leucemia ultrarrápido, altamente preciso y algorítmicamente objetivo que beneficie a los pacientes de todo el mundo. En palabras de Benjamin Bell, citadas por el profesor Haferlach durante un discurso de apertura: “… la IA no sustituirá a los médicos, sino que los potenciará, lo que les permitirá practicar mejor la medicina con mayor precisión y eficacia”.
Conclusión
La misión de MLL es mejorar la atención de los pacientes con leucemia y linfoma a través de diagnósticos de vanguardia. Para ello, el laboratorio utiliza un enfoque interdisciplinario que combina seis disciplinas: citomorfología, inmunofenotipado, análisis cromosómico, hibridación fluorescente in situ (FISH), genética molecular y bioinformática, lo que da como resultado un informe de laboratorio integral y completo.
“En general, podemos distinguir los subtipos de enfermedades a través de métodos fenotípicos como la citomorfología, el inmunofenotipado y las técnicas genéticas. En el futuro, las técnicas moleculares serán los métodos más importantes para ayudarnos a recopilar toda esta información”, dijo el profesor Haferlach. “Hoy, el diagnóstico genético se basa en la citogenética y el análisis de mutaciones. Pronto, haremos la secuenciación del genoma completo y del transcriptoma completo”.
La secuenciación del genoma completo puede identificar más variaciones estructurales y cambios en el número de copias de los que se pueden evaluar a través del análisis cromosómico, y también se puede utilizar para la detección y el análisis de mutaciones, explicó. “Podría haber un ensayo que lo haga todo”, pronosticó. “Si también incluimos perfiles de expresión génica en nuestros diagnósticos, incluso podemos detectar características que ahora están determinadas por inmunofenotipado y citomorfología. Esta es nuestra visión para el futuro”.
“Somos profesores e investigadores”, dijo el Dr. T. Haferlach. “Queremos estar siempre a la vanguardia de lo que viene”. Ese deseo ha llevado a los fundadores de MLL a adoptar e incorporar por completo la tecnología en su laboratorio y a colaborar con AWS para lograr la innovación.
Para obtener más información sobre la genómica en la nube de AWS, consulte aws.amazon.com/health/genomics.
Este artículo fue traducido del Blog de AWS en Inglés