Amazon Web Services ブログ
マルチモーダルなヘルスデータのためのスケーラブルな機械学習パイプラインの構築
この記事は、”Building Scalable Machine Learning Pipelines for Multimodal Health Data on AWS” を翻訳したものです。
ヘルスケアおよびライフサイエンスの組織においては、機械学習(ML)を使用することで、プレシジョンメディシンの実現、患者嗜好の予測、疾患検出、ケアの質の改善などに取り組んでいます。ヘルスケアにおけるITの急速な成長により、ますます多様なデータモダリティから患者レベルのデータが利用可能になりました。さらに、複数のデータドメインからのデータを組み込むことで、MLモデルの有用性と精度を向上させることが研究によって示されています[1]。これは個人のより完全なビューをモデルに与える事によるものだということは、直感的にも理解できます。
マルチモーダル機械学習(マルチモーダルML)と呼ばれる多様なヘルスケアのデータセットにMLを適用することは、研究開発の活発な分野です。ゲノムや医用画像などの多様なデータモダリティからリンクされた患者レベルのデータを分析することで、患者ケアの改善が促進されます。ただし、オンプレミス環境では、単一のモダリティを大規模に分析することは困難でした。また、オンプレミスでの複数のモダリティの非構造化データの処理は、モダリティごとの異なるインフラストラクチャ要件(FPGやGPU要件など)によって、通常困難なものとなります。しかし、AWSを使用すると、専用のパイプラインを簡単にデプロイし、ニーズに合わせて拡張でき、使用した分だけ料金を支払うことができます。
この2部構成のブログ記事では、ヘルスデータに関するマルチモーダルMLのスケーラブルなクラウドアーキテクチャを構築する方法を紹介します。一例として、非小細胞肺がん(NSCLC)のRadiogenomicsデータセットを分析するためのマルチモーダルMLパイプラインをデプロイします。このデータセットには、RNA配列データ、臨床データ(EHRデータより)、医用画像およびその画像のアノテーションから構成されています[2]。このユースケースでは、NSCLCと診断された患者の生存転帰を予測しますが、マルチモーダルMLモデルは、個別治療、臨床診断支援、薬物応答予測などの他のアプリケーションに適用することができます。
この最初のブログ記事では、各データモダリティについて、データ取得、データ処理、および特徴量構築について手順を追って説明します。次のブログ記事では、すべてのデータモダリティから得られた一連の特徴量を使用して患者の生存を予測するモデルを学習し、その結果を1つのモダリティで学習したモデルと比較します。ゲノム、臨床、医用画像データへの応用を紹介しますが、アプローチとアーキテクチャは、幅広いヘルスデータMLユースケースとフレームワークに適用できます。
ソリューションの概要
このアーキテクチャでは、Amazon S3を使用して入力データセットを保存し、パイプラインを使用して各データモダリティを処理および変換し、モデル学習に適した特徴量を作成します(図1を参照)。
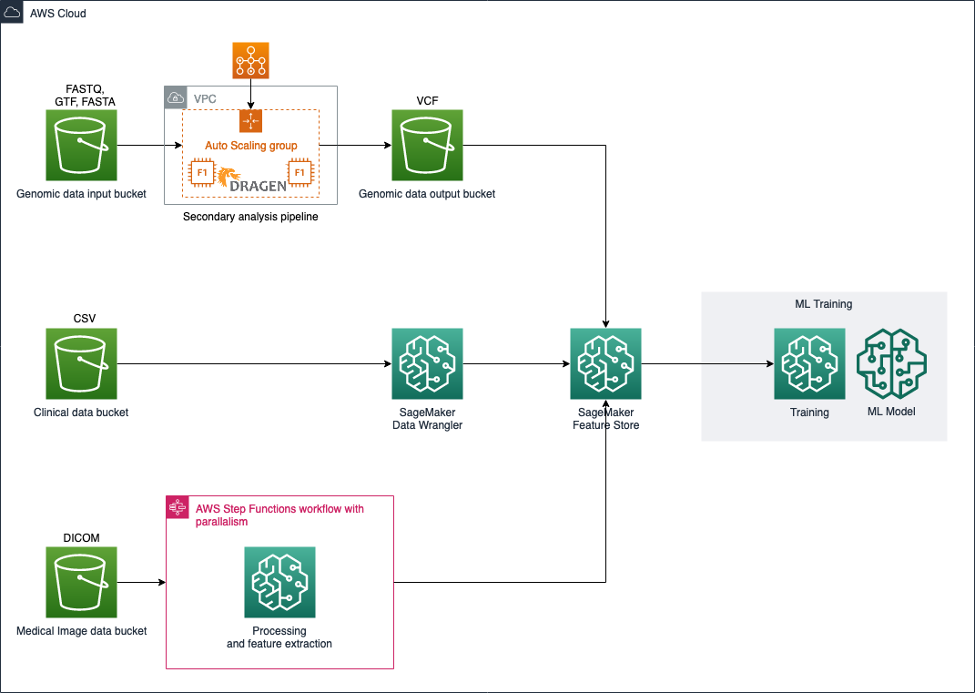
図1: マルチモーダルヘルスデータを統合および分析するためのアーキテクチャ
ゲノムデータについては、Illumina DRAGEN Bio-IT Platform [3]などの二次解析ワークフローをサポートし、DNAやRNAフラグメントのマッピングとアラインメント、バリアントの検出、品質評価、RNAの定量化など、一般的に使用される分析技術が含まれます。NSCLC Radiogenomicデータは、外科的に切除された腫瘍組織のサンプルからのペアエンドRNA配列データで構成されています。S3は、大規模ゲノムデータの分析に適した、安全で耐久性があり、スケーラブルな保管場所です。S3は、シーケンスリードやリファレンスゲノム、中間ファイル、出力データなどを含む入力データを格納するために使用できます。次にシーケンスデータの二次解析を実施し、遺伝子発現レベルを定量化します。出力は表形式で、サンプルあたりの遺伝子ごとの発現レベルが含まれます。個人レベルの遺伝子の定量的な遺伝子発現は、モデル学習の特徴量として使用することができます。
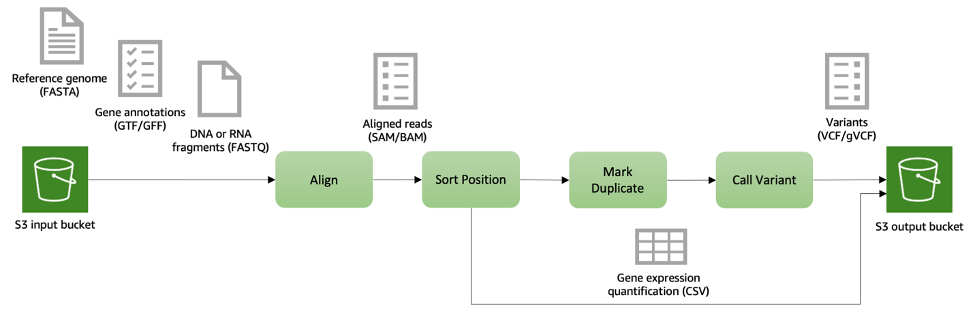
図2: DNAおよびRNAシーケンスデータの二次解析パイプラインの例。入力データ(リファレンスゲノム、遺伝子アノテーション、シーケンスリード)はAmazon S3に格納され、シーケンスアライメント、バリアントコール、遺伝子発現レベルの定量化など様々な処理段階で取得されます。出力ファイルも Amazon S3に保存され、後続の分析において簡単にアクセスできます。
NSCLC Radiogenomicの臨床データは構造化された表形式で、EHRデータや医療保険の請求データと同様です。NSCLCの臨床データには、デモグラフィック(性別、民族)、健康行動(喫煙歴)、がん再発状態、組織病理学的グレード、病理学的TNM分類および生存転帰が含まれます。データ充実化と高度な特徴量エンジニアリングを目的として構築されたインタラクティブツールであるAmazon SageMaker Data Wranglerを活用します。
画像データについては、NSCLC Radiogenomic画像データセット内のCT画像と対応する腫瘍セグメンテーションを使用し、観察された腫瘍の大きさや形状および視覚的属性を示す患者レベルの3次元ラジオミクス特徴量を作成します。
医用画像データは、一般にDICOMファイル形式で格納されます。DICOMはメタデータとピクセルデータを単一のオブジェクトに統合したデータ標準です。肺のCTスキャンなどの3Dスキャンの場合、各断面スライスは通常、個別の DICOMファイルとして保存されます。しかし、MLの目的では、3Dデータを分析したほうが、関心領域(ROI)の全体的なビューが得られるため、より優れた予測が得られます。そのためDICOM形式のスキャンをNiFTI形式に変換します。DICOMファイルをダウンロードしてS3に保存し、Amazon SageMaker Processingを使用してこの変換を実行します。具体的には、各対象ごとにカスタムコンテナを使用してSageMaker Processingジョブを起動し、CTスキャンと腫瘍セグメンテーションの両方の2D DICOMスライスファイルを読み取り、それらを3Dボリュームに結合し、ボリュームをNiFTI形式で保存してS3に書き戻します。
医用画像データを三次元形式で使用して、同じSageMaker Processingジョブの中で腫瘍領域を記述するラジオミクス特徴量を計算します。AWS Step Functionsを使用して、スケーラブルでフォールトトレラントな方法で画像データセット全体の処理を調整します。
最後に、各データモダリティから抽出された特徴量は、ML特徴量を格納するための専用リポジトリであるAmazon SageMaker Feature Storeに書き込まれます。特徴量の準備とモデル学習はAmazon SageMaker Studioを使用して実行されます。

図3: 医用画像処理パイプラインの図。DICOMファイルはAmazon S3バケットに格納されます。処理手順には、2Dスライスから3Dボリュームへの変換、CTとセグメンテーションのアライメント、腫瘍領域内のラジオミクス特徴量抽出、Amazon SageMaker Feature Storeへの特徴量の取り込みが含まれます。
ウォークスルー
前提条件
このウォークスルーの前提条件は次のとおりです。
- Amazon SageMaker、Amazon S3、AWS Step Functions、Amazon EC2、Amazon Athenaをプロビジョニングする権限を持つAWSアカウント。
- 少なくとも1つのパブリックサブネットとNATゲートウェイにルーティングされている1つのプライベートサブネットを持つVPC。
- この例では、次世代シーケンシングデータの二次解析にIllumina DRAGEN Platformを使用します。これには以下が必要です。
- AWSマーケットプレイスでのDRAGEN AMIのサブスクリプション。
- デプロイにおいては、サポートされているAWSリージョンで Amazon EC2のF1インスタンスタイプを選択する必要があります。
- vCPUの制限でf1.4xlargeのEC2インスタンスタイプに、推奨される16のvCPUが許可されていることを確認してください。そうでない場合は、制限の引き上げをリクエストしてください。
- Step Functionsを含む現在の医用画像処理パイプラインは、ml.r5.large インスタンスタイプの50の同時処理ジョブを起動します。アカウントのクォータで50のml.r5.large SageMaker Processingジョブが許可されていることを確認してください。そうでない場合は、制限の引き上げをリクエストしてください。
- このブログに付属するコードリポジトリへのアクセス。
このブログ記事で概説されている各ステップを実行すると、AWSサービスで約13ドルの費用がかかります。
ステップセクションの作成
SageMaker Studio
SageMaker Studioを使用して、データの処理、Jupyter Notebookの利用、およびゲノミクスのパイプラインで使用されるEC2インスタンスへのアクセスが可能です。開始するには、Identity and Access Management (IAM) を使用した標準セットアップの手順に従って、アカウントのSageMaker Studioにアクセスします。簡単のため、図4に示すように、Execution roleのCreate roleを選択し、S3バケットを明示的に指定しないでください。タグ「sageMaker」と値「true」を使用して、SageMakerのS3オブジェクトへのアクセスを許可します。この方法では、S3の入力と出力データのみにアクセスできます。
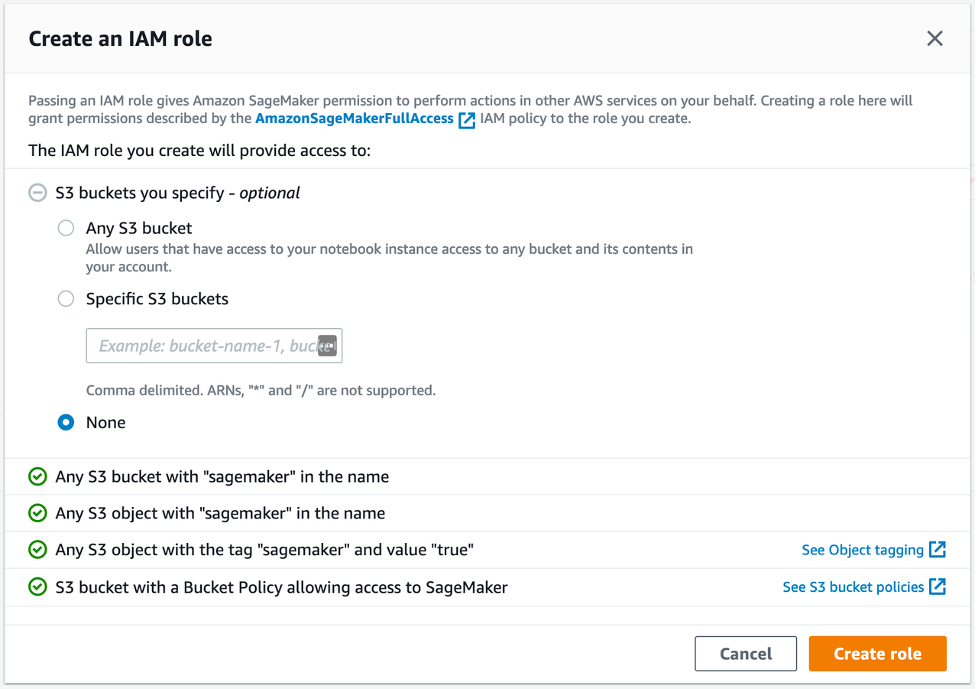
図4: SageMakerのIAMロールの設定
ネットワークのセクションで、VPCでのSageMaker Studioへのアクセスを選択し、プライベートサブネットを指定します。また、AppNetworkAccessTypeを VpcOnlyに設定して、インターネットからの直接アクセスを無効にします。
[送信]を選択してstudioを作成し、環境がプロビジョニングされるまでしばらく待ちます。SageMaker Studio IDEが使用可能になったら、左側のツールバーでGitアイコンを選択し、このブログに付属するリポジトリを複製します。
デフォルトでは、SageMaker Studioのノートブック、ローカルデータ、およびモデルアーティファクトはすべてCustomer Master Key (CMK)で暗号化されます。この例では、匿名化されたデータを扱っています。ただし、Protected Health Information(PHI)を使用する場合、保存中および転送中のすべてのデータを暗号化し、狭義に定義された役割を適用し、最小権限の原則に従ってアクセスを制限することが重要です。ベストプラクティスに関する詳しいガイダンスについては、ホワイトペーパー「HIPAA セキュリティとコンプライアンスの設計」を参照してください。
ゲノムパイプライン
このデモでは、二次解析にIllumina DRAGEN Platformを使用しています。DRAGENは、全ゲノムやRNA、エクソーム、メチローム、がんデータなどの正確で超高速な次世代シークエンシング二次解析パイプラインを提供しています。FPGAベースのAmazon EC2のF1インスタンスを使用して、ゲノム解析アルゴリズムのハードウェアアクセラレーションの実装が可能です。DRAGEN RNA パイプラインを実行して、リードをヒトhg19リファレンスゲノム [2] にマッピングします。また、遺伝子アノテーションファイル(GTF/GFF形式)を使用して、リードをスプライスジャンクションにアラインさせ、遺伝子発現レベルを定量化します。hg19ゲノムのGTFファイルは、GENCODE プロジェクトからダウンロードできます。DRAGEN RNAパイプラインを実行する手順は次のとおりです。
- AWSマーケットプレイスで DRAGEN AMIをサブスクライブ
- AMIを使用して、SageMaker Studioに使用したのと同じVPCおよびプライベートサブネットで新しいf1.4xlargeインスタンスを起動する
- ストレージにデフォルトの100GBのAmazon EBS gp2ボリュームを使用する
- タグを追加する手順をスキップする
- プライベートサブネット内からのすべてのTCPトラフィックを許可するセキュリティグループを使用する
新しく起動されたインスタンスにアクセスするには、[ファイルのアップロード]アイコンをクリックしてEC2キーペアをSageMaker Studioにアップロードし、[ファイル]メニューから新しいターミナルを起動します。次に、SageMaker StudioターミナルからEC2インスタンスのプライベートIPアドレスにSSH接続します。
RNA配列データはSRA(シーケンス・リード・アーカイブ)データとしてアクセスできます。SRAファイルは、Open DataのRegistryを通じてAWSでホストされ、パブリックにアクセスできます。ペアエンドリードの場合、「.1」と「.2」の読み込みを含む2つのFASTQファイルが生成されます。RNA配列データの詳細は、米国国立生物工学情報センター (NCBI) [4]で確認できます。SRAファイルをダウンロードし、SRAツールからfastq-dumpを使用して、EC2インスタンスで以下のコマンドを実行してFASTQ形式に変換します。
aws s3 cp s3://sra-pub-run-odp/sra/<ACCESSION-ID>/<ACCESSION-ID> <ACCESSION-ID.sra>
fastq-dump -I --split-files <ACCESSION-ID.sra>
ヒトhg19リファレンスゲノムをEC2インスタンスにダウンロードし、DRAGEN RNAパイプラインに固有のリファレンスハッシュテーブルを作成します。
aws s3 cp s3://ngi-igenomes/igenomes/Homo_sapiens/UCSC/hg19/Sequence/WholeGenomeFasta/genome.fa hg19.fa
dragen --build-hash-table true --ht-build-rna-hashtable true --ht-reference hg19.fa --output-directory <HASHTABLE DIRECTORY>
FASTQファイル、リファレンスハッシュテーブル、およびGTFファイルを使用して、DRAGEN RNAパイプラインの遺伝子発現定量化モジュールを実行します。
dragen --enable-rna=true --enable-duplicate-marking=true --enable-rna-quantification=true --enable-bam-indexing=true --alt-aware=true --output-format=bam --enable-map-align-output=true --enable-map-align=true --enable-sort=true --annotation-file=<GTF FILE > --ref-dir=<HASHTABLE DIRECTORY> --output-directory=. --output-file-prefix=<PREFIX> --input-qname-suffix-delimiter=. -1 <FASTQ_FILE READ1> -2 <FASTQ_FILE READ2> --RGID <ID> --RGSM <ID>
このデモにはAWS MarketplaceのDRAGEN AMIを使用しましたが、本番規模のデータ分析では、DRAGENクイックスタート [5] を利用できます。AWSコマンドラインインターフェイスとAWS Batchコンソールの両方からジョブを実行できます。
上記のコマンドは、サンプルあたりの遺伝子ごとの発現レベルを表形式で出力ファイルを生成します。図5に示すように、各行は症例IDまたは患者に対応し、各列は特定の遺伝子の定量化された発現レベルを表します。この定量化結果は、マルチモーダルMLモデルを学習するための特徴ベクトルとして使用されます。

図5: RNA配列パイプラインによって生成された出力ファイル。各患者(行)の遺伝子(列)の発現レベルを定量化している。
CSV形式で定量化された遺伝子発現レベルを含むこの出力ファイルをS3に格納します。
aws s3 cp <OUTPUT-FILE> <S3-BUCKET-GENOMIC-DATA>
臨床パイプライン
匿名化された臨床データは、Cancer Imaging Archive (TCIA) [6]から直接CSV形式でダウンロードし、以下に示すようにS3に保存することができます。
curl -o Clinical-Data.csv https://wiki.cancerimagingarchive.net/download/attachments/28672347/NSCLCR01Radiogenomic_DATA_LABELS_2018-05-22_1500-shifted.csv
aws s3 cp Clinical-Data.csv <S3-BUCKET-CLINICAL-DATA>
SageMaker Studio内から新しいデータフローを作成して、データを取り込みます。S3からデータをインポートするオプションを選択し、臨床データファイルを選択し、データセットをインポートします。
SageMaker Data Wranglerを使用して、臨床データの前処理を行っています。サンプルデータの準備は最小限で済むので、SageMaker Data Wranglerの多数の機能のほんの一部を使用します。つまり、SageMaker Clarifyを使用してジェンダーバイアスなどのデータバイアスを特定し(図6)、ターゲット変数の生存状況に関するデータリークを探索し(図7)、特徴量変換を行います。
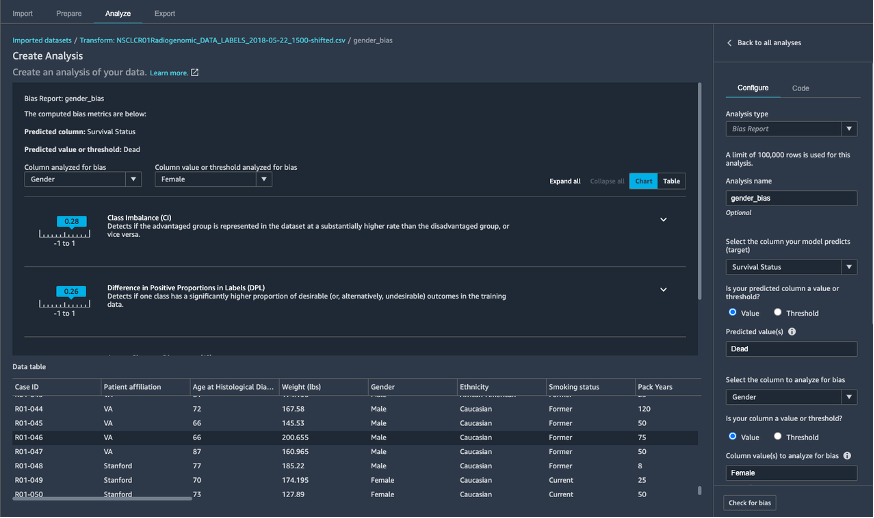
図6: Amazon SageMaker Data Wranglerに組み込まれているSageMaker Clarifyを使用したデータセットのジェンダーバイアスの理解
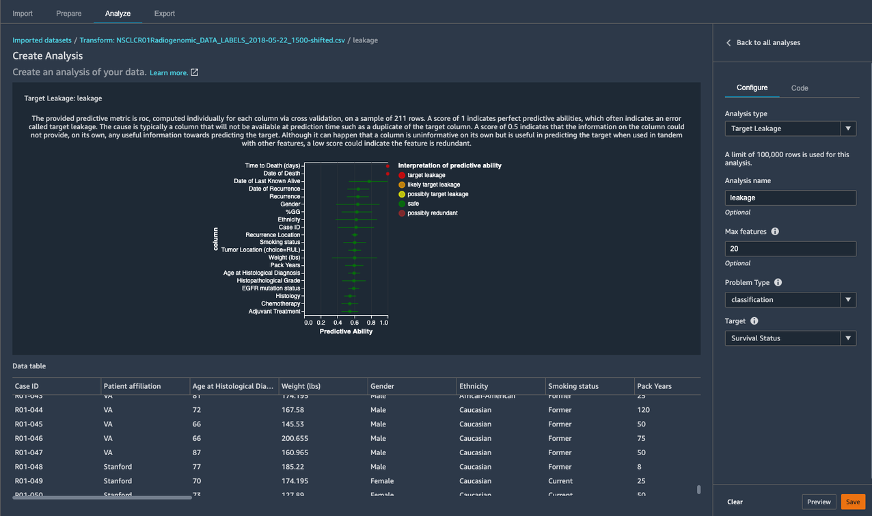
図7: SageMaker Data Wranglerでターゲットリーク解析を使用して特徴量にデータリークがあるかないかを理解
![Interactively encoding the Survival Status column from string to [0, 1] in SageMaker Data Wrangler](https://d2908q01vomqb2.cloudfront.net/b3f0c7f6bb763af1be91d9e74eabfeb199dc1f1f/2021/07/21/Interactively-encoding-the-Survival-Status-column-from-string-to-0-1-in-SageMaker-Data-Wrangler.png)
図8: SageMaker Data WranglerのSurvival Status列を文字列から[0, 1]にインタラクティブにエンコード
SageMaker Data Wrangler内で以下の変換をデータに適用します。
- Amazon SageMaker Transform Dataで説明されているように、すべてのカテゴリ列でワンホットエンコードする
- ターゲットリークを含み、画像日付に関連する列を削除する
- ターゲット列「Survival Status」をエンコードする(図8)
- カスタム変換を使用して、[Weight (lbs)]または[Pack Years]列の値が収集されない行を削除する
- Handle Missingを使用して欠損値をゼロで埋める
- SageMaker Feature Storeでバージョン管理のために必要に応じてイベント時間列を追加する
- イベント時間列を追加するには、[準備] タブで [変換]を選択し、新しい変換をカスタム式として定義します。式は、タイムスタンプのSpark SQL式、つまりcurrent_timestamp()で、出力列名をタイムスタンプに設定します。これにより、データセットに列が追加され、各行にタイムスタンプが格納されます。
次に、[エクスポート]タブを選択し、Data Wranglerフローに従って、特徴量をFeature Storeにエクスポートします。SageMakerは、エクスポートジョブを処理するためにJupyterノートブックを自動的に作成します。ただし、ノートブックを実行する前に、いくつかの変更を行う必要があります。以下に示す既存のセルを変更して、record_identifier_name変数と event_time_feature_name変数をデータの対応する列名に割り当てます。
record_identifier_name = "Case_ID"
if record_identifier_name is None:
raise RuntimeError("Select a column name as the feature group identifier.")
event_time_feature_name = "timestamp"
if event_time_feature_name is None:
raise RuntimeError("Select a column name as the event time feature name.")
この例では、モデルトレーニングに焦点を当てているため、オフラインFeature Storeのみを使用します。既存のセルを次のように更新して、オンラインストアを無効にします。
# controls if online store is enabled. Enabling the online store allows quick access to
# the latest value for a Record via the GetRecord API.
enable_online_store = False
次に、ノートブック内のすべてのセルを実行し、処理が完了すると、処理された特徴量のFeature Storeの場所を示す出力が表示されます(図9)。
'ProcessingOutputConfig': {'Outputs': [{'OutputName': '6e72c81c-d6c7-41bc-9557-e20e93ce2af5.default', 'FeatureStoreOutput': {'FeatureGroupName': 'clincal-feature-group-xx-xxx-xxxx-xxxx'}, 'AppManaged': True}]}

図9: 処理された特徴量はFeature StoreタブのFeature groupのリストに示すように、Feature groupに格納される
医用画像パイプライン
データダウンロード
医用画像データのダウンロードには「.tcia」マニフェストファイルが必要です。このファイルは、Cancer Imaging Archive (TCIA) [6]からダウンロードできます。Linuxと親和性のあるNBIA データ取得 CLI ツールを使用して、SageMaker Studioにアタッチされたボリュームに画像データをダウンロードします。まず、データ取得ツールをダウンロードしてビルドします。
cd SageMaker
sudo yum install golang
git clone https://github.com/ygidtu/NBIA_data_retriever_CLI.git ./data_retriever
cd data_retriever/
chmod +x build.sh
./build.sh
次に、データ取得ツールを実行して、マニフェストファイルに記述されているすべてのデータをSageMaker Studioにアタッチされたボリュームにダウンロードします。
mkdir data
cd data
../data_retriever/nbia_cli_linux_amd64 -i <path to manifest file>NSCLC_Radiogenomics-11-10-2020Version3.tcia -o . -p 3
データ取得ツールはディレクトリnsclc_radiogenomicsを作成し、その内部に、対応する医用画像データを含む各患者のサブディレクトリを作成します。特定のスタディは、1つ以上の画像シリーズのディレクトリと、各画像シリーズを説明するメタデータを持つjsonファイルで構成されます。画像シリーズディレクトリには、CT/PETスキャンの1つ以上の2D DICOMファイル、または腫瘍セグメンテーション用の単一のDICOMファイルがあります。腫瘍セグメンテーションDICOMファイルは、ソフトウェアによって作成され、胸部放射線科医によってレビューされた3Dセグメンテーションオブジェクトで構成されています。
ダウンロードが完了したら、AWS CLIを使用してS3バケットにデータをコピーします。
aws s3 mb s3://multimodal-image-data/
aws s3 sync ./nsclc_radiogenomics/ s3://multimodal-image-data/nsclc_radiogenomics/
S3のデータを使用して、必要な画像データ処理とMLを大規模に実行する準備が整いました。
DICOM処理
この例では、腫瘍セグメンテーションを伴うCTスキャンのみに焦点を当てています。したがって、患者およびスタディごとに、jsonメタデータファイルを読んで、シリーズにスキャンイメージングシリーズとセグメンテーションオブジェクトの両方が含まれているかどうかを判断します。その場合は、2D DICOMファイルのセットとしてダウンロードされた各CTスキャンイメージングシリーズを、単一の3D NIFTIファイルに変換します。次に、dcmstackというPythonパッケージを使用してDICOMからNIfTIへの変換を実行し、すべての DICOMファイルを読み込み、空間スライスの位置に従ってソートし、スライスをスタックして3Dボリュームを作成します。3DボリュームはNiBabel pythonパッケージでNiFTI形式で書き出されます。腫瘍セグメンテーションDICOMオブジェクトごとに、Pydicomパッケージを使用して3Dアレイを読み込み、対応するCTスキャンのボリュームに合わせてボリュームの向きを変え、出力をNiFTIファイルとして保存します。この完全なプロセスは、リポジトリのPythonスクリプト imaging/src/dcm2nifti_processing.pyに書かれています。
一部のスタディのセグメンテーションオブジェクトは、空のスライスが切り取られたDICOMオブジェクトに保存されていることに注意してください。これにより、CTスキャンのスライスの数と対応するセグメンテーションオブジェクトの間で不一致が発生します。これに対処するには、ImagePositionPatient DICOM属性の値を一致させて、腫瘍セグメンテーションをCTスキャンの対応する位置に揃え、セグメントをゼロで埋めて、同じ数のスライスを持つようにします。このプロセスはimaging/src/dcm2nifti_processing.pyスクリプトにも実装されています。
図10は、CTスキャンで腫瘍マスクを黄色で透明に重ねた例を示しています。(ケースID R01-093)
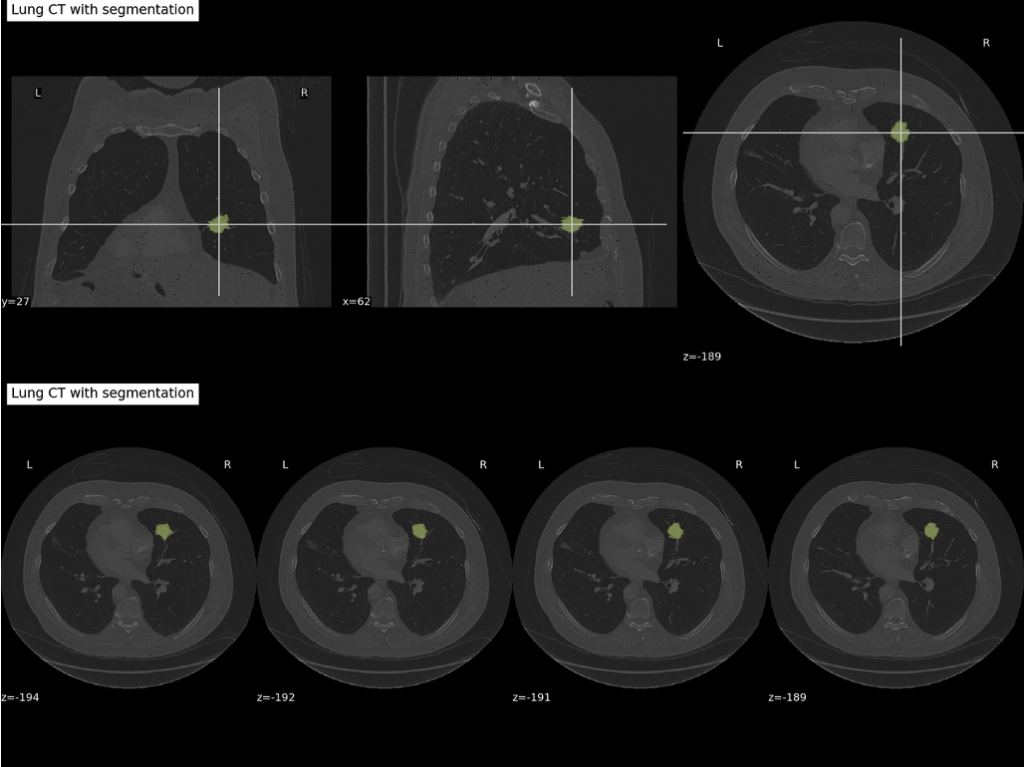
図10:肺腫瘍マスクを黄色に重ねたCTスキャンの可視化例
ラジオミクス特徴量抽出
アノテーション付きCTスキャンにおける腫瘍を記述するラジオミクス特徴量をpyradiomics [7]を使用して計算していきます。ライブラリを使用して、腫瘍の関心領域内の強度の分布と共起の統計的表現、腫瘍を形態学的に記述した形状の測定など、8つのクラスの120のラジオミクス特徴量を抽出します。ラジオミクス特徴量の計算は、imaging/src/radiomics_utils.pyスクリプトの compute_features関数のradiomicsFeatureExtractorクラスに変換されたNIfTIイメージを提供することによって、ボリューム的に実行されます。
特徴量が計算されると、後の「マルチモーダル特徴量ストア」セクションで説明されているように、これらの特徴量はプログラムによってSageMaker Feature Storeに書き込まれます。
Step FunctionsとSageMaker Processingによるスケーリング
2つのPythonスクリプト imaging/src/dcm2nifti_processing.pyと imaging/src/radiomics_utils.py を使用して、データセットにスタディIDを指定して1つのスタディを処理できます。臨床研究で一般的に見られるように、数百または数千のスタディにスケールするには、スクリプトのスケーラブルで伸縮性のある実行のためにSageMaker Processingを使用し、AWS Step Functionsをスタディの並列処理のためのオーケストレーションレイヤーとして使用します。
SageMaker Processingは、完全マネージドインフラストラクチャ上のDocker コンテナイメージでコードを実行します。ここで、上記の2つのPythonスクリプトと、SageMaker Studioでsagemaker-studio-image-buildを使用して (requirements.txtファイルで指定) ランタイム要件を含む image/src/DockerFileで定義されたDockerコンテナイメージをビルドします。次のコードスニペットは、imaging/preprocess-imaging-data.ipynbにあります。
cd src/
!sm-docker build . --repository medical-image-processing-smstudio:1.0
このコマンドの最後に、Dockerコンテナイメージが構築され、AWS Elastic Container Registryにプッシュされます。
Image URI: <account-id>.dkr.ecr.<region>.amazonaws.com/medical-image-processing-smstudio:1.0
次に、Step FunctionsステートマシンでSageMaker Processingジョブを指定して、1つのDICOMスタディを処理するのに十分なRAMとディスク容量がある5 GBのディスクボリュームを持つml.r5.largeのインスタンスを1つ使用します。
Step Functionsステートマシンの設計を図11に示します。ステートマシンの定義は、リポジトリ内のnsclc-radiogenomics-imaging-workflow.jsonにあります。

図11: AWS Step Functionsコンソールでレンダリングされたステートマシンのワークフロー
Map状態を設計して、アレイの複数のエントリ、つまり各イメージングスタディに対して同じステップをステート入力で実行します。同時処理ジョブの最大数を定義するMap状態でmaxConcurrencyを50に設定します。ステートマシンをフォールトトレラントにするには、エラー処理Catchフィールドを追加して、DICOM/NIfTI 変換内のエラーをフォールバックステップにルーティングします。これにより、1 つ以上のスタディで DICOM スタディ/セグメンテーションの破損など、予期しないエラーによる実行全体の失敗を回避できます。もう1つのフォールトトレラントなメカニズムは、StepFunctions が50の同時SageMaker プロセッシングジョブを送信したときに起動時に発生する可能性がある ThrottlingException (レート超過)を処理するためのバックオフを使用した再試行ロジックです。
imaging/preprocess-imaging-data.ipynbの次のコードスニペットは、すべてのスタディのワークフローを実行し、ラジオミクス特徴量を指定された特徴量ストアに保存します。
import boto3
import json
import uuid
sfn = boto3.client('stepfunctions')
stateMachineArn = 'arn:aws:states:<region>:<accountId>:stateMachine:nsclc-radiogenomics-imaging-workflow'
suffix = uuid.uuid1().hex
payload = {
"PreprocessingJobName": "dcm-nifti-conversion-%s" % suffix,
"FeatureStoreName": "nsclc-radiogenomics-imaging-feature-group",
"OfflineStoreS3Uri": "s3://<S3-BUCKET-IMAGE-DATA-PROCESSED>/nsclc_radiogenomics-multimodal-imaging-featurestore",
"Subject": [
"R01-001", "R01-002", ...., "R01-162", "R01-163"
]
}
sfn.start_execution(stateMachineArn=stateMachineArn, name=suffix, input=json.dumps(payload))
ひとつの症例の変換と特徴量抽出には、約5分かかります。MaxConcurency = 50で実行すると、データセット全体の処理を162 * 5 = 800分ではなくわずか 20分で完了します。
環境削除
今後の料金が発生しないようにするには、上記の手順で作成したリソース (EC2インスタンス、SageMakerサービス、S3上のデータ、Amazon ECRのコンテナ、Step Functions、AWS KMSキー)を削除します。ただし、この2部構成のブログシリーズの第2部を引き続き実施する予定がある場合を除きます。
まとめ
このブログでは、一連のデータ分析パイプラインを展開して、多様な非構造化データモダリティからデータを効率的に処理する方法を説明しました。フルマネージド型のAWSサービスを活用することで、マルチモーダルデータを大規模に処理するためのセットアップを効率化しました。
この2部構成のシリーズの次のブログ記事では、SageMaker Feature Storeを使用してマルチモーダルフィーチャストアを構築します。次に、各データモダリティの特徴量に基づいてモデルを個別に学習させ、一緒にプールされたすべてのモダリティでモデルを学習させます。我々は、それぞれのモデルの性能を比較し、マルチモーダルモデルが各モダリティからの情報をどのように使用して予測を行うかを定量化します。
マルチモーダルデータを活用することで、ヘルスケアおよびライフサイエンス向けのより良いMLモデルが得られ、その後のケア提供と患者の転帰が改善されます。次のブログ記事を引き続きフォローし、またここで紹介した例を試してみることをお勧めします。
AWSのヘルスケアとライフサイエンスに関する詳細については、aws.amazon.com/healthをご覧ください。
参考資料
[1] Huang, Shih-Cheng, et al. “Fusion of medical imaging and electronic health records using deep learning: a systematic review and implementation guidelines.” NPJ digital medicine3.1 (2020): 1-9.
[2] Bakr, Shaimaa, et al. “A radiogenomic dataset of non-small cell lung cancer.” Scientific data 5.1 (2018): 1-9.
[3] www.illumina.com/dragen
[4] Gene Expression Omnibus: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE103584
[5] https://aws-quickstart.github.io/quickstart-illumina-dragen/
[6] Bakr, S. et al. The Cancer Imaging Archive http://doi.org/10.7937/K9/TCIA.2017.7hs46erv (2017).
[7] Griethuysen, J. J. M, et. al. Computational Radiomics System to Decode the Radiographic Phenotype. Cancer Research (2017)
著者について
Olivia Choudhury
Olivia Choudhury, PhDはAWSのパートナーソリューションアーキテクトです。彼女は、ヘルスケアおよびライフサイエンス分野のパートナーの、AWS を活用した最先端ソリューションの設計、開発、拡張を支援しています。彼女は、ゲノミクス、ヘルスケア分析、連合学習、プライバシー保護機械学習のバックグラウンドを持っています。仕事以外では、ボードゲームをしたり、風景を描き、漫画を集めています。
Andy Schuetz
Andy Schuetz, PhD はAWSのシニアスタートアップソリューションアーキテクトで、お客様がヘルスケアおよびライフサイエンスソリューションを利用できるよう支援することに注力しています。AndyがAWSで構築作業をしていないとき、彼はマウンテンバイクに乗ることが好きです。
Michael Hsieh
Michael Hsiehは、シニアAI/MLスペシャリストソリューションアーキテクトです。彼はHCLSのお客様と協力して、AWSテクノロジーと医用画像処理に関する専門知識を活用してMLジャーニーを進めています。シアトルの住民として、彼はハイキングコース、SLUでのカヤック、シルスホール湾の夕日など、街が提供する偉大な母なる自然を探索するのが大好きです。
翻訳はIndustry Solutions Architectの松永が担当しました。原文はこちらです。