AWS Public Sector Blog
Accelerating genome assembly with AWS Graviton2
One of the biggest scientific achievements of the twenty-first century was the completion of the Human Genome Project and the publication of a draft human genome. The project took over 13 years to complete and remains one of the largest private-public international collaborations ever. Advances since in sequencing technologies, computational hardware, and novel algorithms reduced the time it takes to produce a human genome assembly to only a few days, at a fraction of the cost. This made using the human genome draft for precision and personalized medicine more achievable.
Researchers derived the international reference genome routinely used in human genomics workloads from the sequence of a few different anonymous individuals, with the majority of the genome (70 percent) derived from a single individual. This hampers the use of the reference genome for clinical purposes, given natural genetic differences in various subpopulations. For true personalized, precision medicine, there is an urgent need to develop more genome assemblies specific to various subpopulations, perhaps even at an individual level. To enable this, several efforts around the world are working on developing draft human genome assemblies. In the US, NIH’s Human Pan Genome Project (HPRC) is generating sequence reads from various publicly available human samples. These reads are then deposited in Amazon Simple Storage Service (Amazon S3) and available for researchers to download and use for research through the Amazon Web Services (AWS) Registry of Open Data.
In a previous blog, we introduced a generalized benchmarking framework to test genomics workloads across a wide variety of AWS instance types. We also demonstrated how AWS Graviton2 instances provide increased cost and performance efficiencies for a routine task. In this blog, we demonstrate how to do a genome assembly in the cloud in a cost-efficient manner using ARM-based AWS Graviton2 instances. The original Peregrine genome assembler algorithm (Human Genome Assembly in 100 Minutes) was rewritten using the Rust programming language. The rewrite provides a unique opportunity to test out generating genome assemblies on both ARM-based Graviton2 instances and Intel CPUinstances setup with AWS Batch.
Public Peregrine2021 (v0.2.2 beta) docker images are available via the AWS public container registry.
Graviton2: public.ecr.aws/t1i7k1n6/assembly/rpg-test,
Intel x86_64: public.ecr.aws/t1i7k1n6/assembly/rpg-x86_64-test
The public docker images are built for use with the Amazon Elastic Container Service (Amazon ECS) and AWS Batch. We utilize an Amazon Elastic Compute Cloud (Amazon EC2) Launch Template to launch the host computing node through an Amazon EC2 Autoscaling Group. The Terraform launch template sets up enough scratch space preparing for the extensive input data, large intermediate files, and the genome assembly results. Once the genome assembly farm is built, we can submit jobs from the AWS Batch console or AWS Command Line Interface (AWS CLI).
AWS CLI Job submission:
aws batch submit-job --job-name ecoli-test --job-queue asm-aarch-64 --job-definition asm-64cores --parameter prefix=<s3://asm-test/test-ecoli>
Here is an example (Figure 1) running a human genome assembly within two hours, including fetching the source data, using an r6gd.16xlarge instance on AWS. The assembly results are then deposited into a pre-specified Amazon Simple Storage Service (Amazon S3) location.
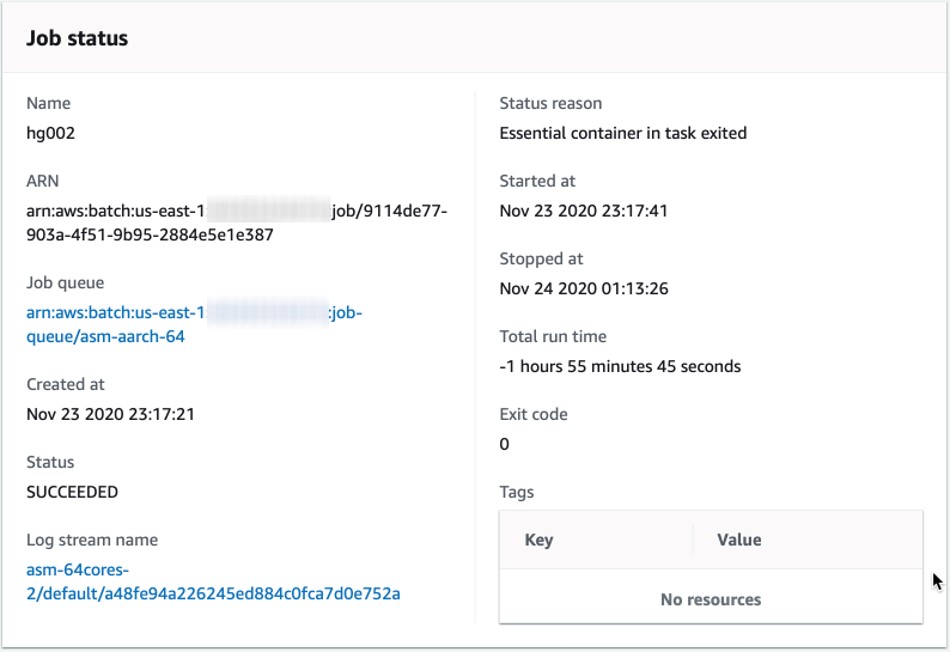
Figure 1: Results from running a human genome assembly
Benchmarking genome assembly using ARM Graviton2 instances
Table 1 Results from benchmarking assembly run across multiple AWS instances. ‘hg-total-cost’ refers to the costs from running the HG002 assembly, ‘jlm-total-cost; refers to the costs from running the Jamiacan Lion Mother assembly.
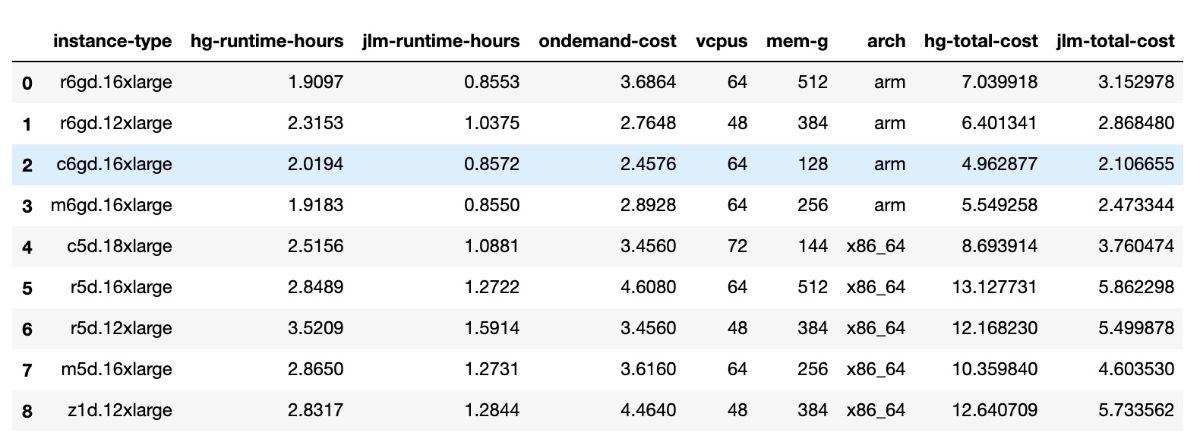
Table 1: Results from benchmarking assembly run across multiple AWS instances. ‘hg-total-cost’ refers to the costs from running the HG002 assembly, ‘jlm-total-cost; refers to the costs from running the Jamiacan Lion Mother assembly.
Human genome assembly
To test the speed and cost efficiencies that can be achieved using the ARM instances, we ran the HG002 assembly with the Data Freeze v1.0 set (https://github.com/human-pangenomics/HG002_Data_Freeze_v1.0, ~34x genomic coverage) on a few instance types. The results are described in the Table 1 and Figure 2.

Figure 2: Benchmarking results, comparing total-cost on the y-axis and vcpus on x-axis. The panel on the left shows the results from the human genome (HG002). Panel on the right shows results from the Jamaican-Lion-Mother assembly.
From the results, we can conclude:
- In our results, ARM offers the best choice for both performance and cost. For comparable instance types (ARM vs x86_64), we see an average of 50 percent increased runtime for x86_64 when compared with similar ARM instance types. This results in an increase of 85-90 percent increase in cost.
- Increasing total memory did not sufficiently reduce the runtime to justify the increased cost of those instances. Because of this, it looks like the compute optimized instances performed better in terms of cost/performance (the C6/C5 family of instances produced the lowest cost run). The C6 instances were not the fastest of the ARM instances tested, but it did come in as the cheapest.
Cannabis genome assembly
Beyond using de novo genome assembly for the human genome, getting complete genome assemblies is extremely important for agricultural biotech applications. Unlike the human genome, plant genomes are not as well studied, have a large range of genome sizes (from a couple hundred megabases to 10s of gigabases) and very different repeat content that makes plant genome assembly a very difficult yet important challenge to solve. We chose to test the assembler performance on AWS using some long read sequencing data that was available for the cannabis genome. Cannabis is one of the oldest cultivated crops, used as a source of food, fibre, and medicine. Possibly due to scientific inquiry restrictions due to drug policy, the taxonomy and evolutionary history is largely unknown. Advances in understanding the cannabis genome may help scientific understanding of its role in both clinical settings and as an important agricultural product. The cannabis assembly results (Table 1 and Figure 2) were similar when compared with the HG002 assembly. ARM instances provide a significant advantage in terms of performance and cost. Instance types with more memory do not provide a significant decrease in runtime to justify the additional cost.
Summary
The assembly results may vary depending on the parameters used. With the Peregrine2021 v0.2.2, we get an assembly size ~5.53Gb (from both haplotype of the HG002 genome). The total size from primary contigs is ~3.24G with a N50 ~40Mb). For the cannabis genome (Jamaican Lion), we get an assembly of size ~1.13G with a N50 of ~3.25Mb.
How many genomes can we assemble in one day?
For our test account, the standard limit of AWS vCPUs by default is 512 cores, which equates to eight c6gd.16xlarge with 64 vCPUs can be launched at once. Assuming each genome assembly takes around two hours (Table 1), a user should be able to run 96 similar genome assemblies in a day.
Why is this important?
The advent of long-read sequencing technologies (Pacific Biosciences and Oxford Nanopore) revolutionized the field of human disease genetics. The first human genome assembly done with long-read DNA sequencing data took 400,000+ CPU hours in 2014. Since then, technological advances in sequencing technologies and novel algorithm development tremendously improved the efficiency of the process. As demonstrated with this blog, AWS Batch and AWS Graviton2 infrastructure has the ability to assemble a genome in two hours. This would prove revolutionary and will open many new opportunities to study human diseases in a more comprehensive matter. We believe such efficient computation tools and cloud infrastructure will be the foundation for resolving challenges of healthcare and agriculture in the twenty-first century.
Learn more about how AWS is supporting biomedical research
For more information on how AWS helps solve complex research workloads and enables scientific research, see the AWS Research and Technical Computing webpage and access our library of AWS Education: Research Seminars. For NIH funded research projects, request more information about the NIH STRIDES Initiative.